Fibrosi cistica
Che cos'è la fibrosi cistica?
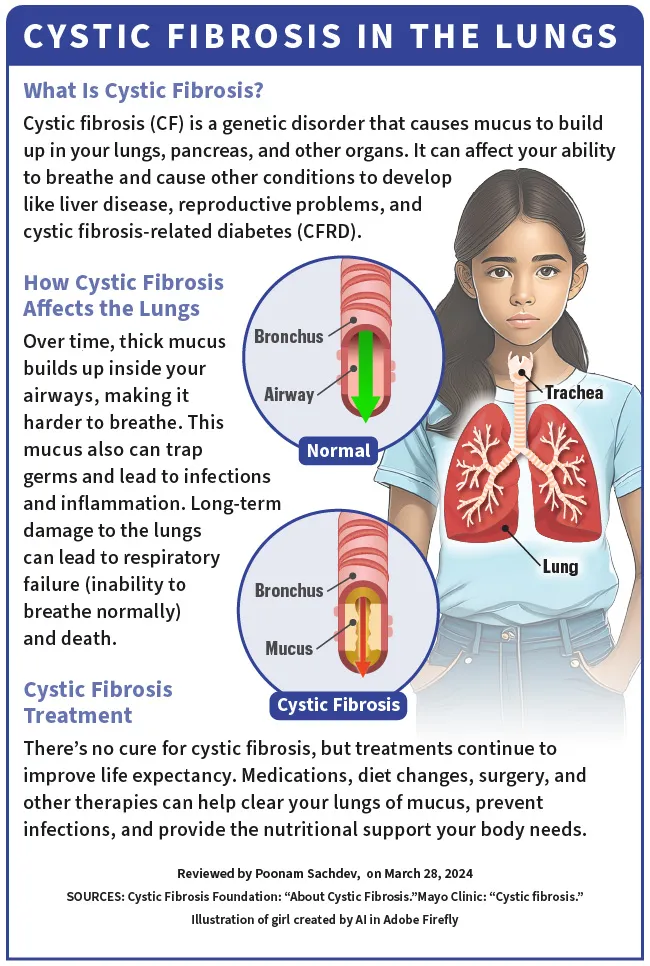
La fibrosi cistica (FC) è una malattia genetica, il che significa che la si trasmette dai genitori alla nascita. Colpisce i polmoni, il pancreas e altri organi. La CF cambia il modo in cui il cloruro (sale) si muove attraverso le cellule del corpo. Ciò fa sì che il muco (che dovrebbe essere sottile e scivoloso) in vari organi diventi denso e appiccicoso.

Con il tempo, questo muco denso si accumula all'interno delle vie respiratorie, rendendo difficile la respirazione. . Il muco intrappola i germi e porta a infezioni e infiammazioni. Può anche causare gravi danni a lungo termine ai polmoni e portare a insufficienza respiratoria (incapacità di respirare normalmente) e morte.
Nel pancreas, il muco denso causato dalla fibrosi cistica impedisce il rilascio di enzimi digestivi quando si mangia. Ciò porta alla malnutrizione e alla scarsa crescita. La fibrosi cistica può anche causare malattie del fegato, problemi riproduttivi e diabete correlato alla fibrosi cistica (CFRD).
Più di 40.000 persone negli Stati Uniti vivono con la fibrosi cistica. I medici diagnosticano circa 1.000 nuovi casi ogni anno. Oggi, più della metà della popolazione affetta da fibrosi cistica ha almeno 18 anni e i nuovi trattamenti hanno allungato di decenni l'aspettativa di vita.
Sintomi della fibrosi cistica
Le persone affette da fibrosi cistica possono avere sintomi che includono:
Fibrosi cistica atipica
Esiste anche una forma di fibrosi cistica chiamata "fibrosi cistica atipica". È un tipo più lieve e può colpire solo un organo. I sintomi di solito si manifestano molto più tardi nella vita rispetto alle persone con fibrosi cistica tipica. A differenza della fibrosi cistica, non esiste una definizione standard di fibrosi cistica atipica. I sintomi della fibrosi cistica atipica possono includere:
Cause della fibrosi cistica
La fibrosi cistica è causata da un cambiamento, o mutazione, in un gene chiamato CFTR (regolatore della conduttanza transmembrana della fibrosi cistica). La proteina contenuta in questo gene controlla il flusso di sale e liquidi dentro e fuori le cellule. Se il gene CFTR non funziona correttamente, nel tuo corpo si accumula muco appiccicoso.
Per contrarre la fibrosi cistica, devi ereditare la copia mutata del gene da entrambi i tuoi genitori . Sono note più di 1.700 mutazioni del gene CFTR. Il novanta per cento delle persone colpite ha almeno una copia della mutazione F508del.
Se erediti una sola copia, non avrai alcun sintomo, ma sarai portatore della malattia. Ciò significa che esiste la possibilità che tu possa trasmetterlo ai tuoi figli.
Circa 10 milioni di americani sono portatori di FC. Ogni volta che due portatori di FC hanno un bambino, c'è una probabilità del 25% (1 su 4) che il loro bambino nasca con FC.
Diagnosi della fibrosi cistica
La diagnosi precoce significa un trattamento precoce e una salute migliore più avanti nella vita. Ogni stato degli Stati Uniti testa i neonati per la fibrosi cistica utilizzando uno o più di questi tre test:
Esame del sangue. Questo test controlla i livelli di tripsinogeno immunoreattivo (IRT). Le persone affette da FC ne hanno livelli più elevati nel sangue. Ogni stato esegue almeno un esame del sangue per lo screening neonatale.
Test del DNA. Cerca mutazioni nel gene CFTR.
Test del sudore. Questo test indolore misura il sale (cloruro) nel sudore. Se i tuoi risultati sono più alti del normale, suggerisce CF.
La diagnosi della fibrosi cistica prevede diversi passaggi. Una valutazione completa dovrebbe includere uno screening neonatale, un test del cloruro nel sudore, un test genetico o del portatore (DNA) e una valutazione clinica presso un centro di cura accreditato.
La maggior parte delle persone affette da FC sono diagnosticata all'età di 2 anni. Ad alcune persone che non sono state sottoposte al test alla nascita non viene diagnosticata la fibrosi cistica finché non diventano adulte. Il tuo medico potrebbe prescriverti test del DNA o del sudore se hai sintomi di fibrosi cistica.
Un test del cloruro nel sudore è il modo più affidabile per diagnosticare la fibrosi cistica.
Se il tuo bambino ha un esame del sangue che indica fibrosi cistica ma ha un risultato intermedio (inconcludente ) test del sudore, il medico può diagnosticare al bambino la sindrome metabolica correlata al CFTR (CRMS). Le prospettive di una persona affetta da CRMS non sono chiare, ma potrebbe avere un rischio maggiore di problemi alle vie aeree, ai seni, al sistema riproduttivo, all'intestino o al pancreas.
Se un test del sudore o un test genetico non è conclusivo, altri due test possono aiutare a diagnosticare la fibrosi cistica:
Differenza potenziale nasale (NPD). Si implica il passaggio di una piccola corrente elettrica attraverso il rivestimento nasale (epitelio). Vengono applicate diverse soluzioni al rivestimento nasale e viene misurata la corrente elettrica. Le persone con FC rispondono a questo test in modo molto diverso rispetto a quelle senza FC.
Misurazione della corrente intestinale (ICM). Implica una biopsia indolore (test di laboratorio di tessuto campione) del tessuto rettale per testare la funzione CFTR delle cellule.
Sebbene gli esami del sangue, del DNA e del sudore siano i metodi più comuni e affidabili per diagnosticare la fibrosi cistica, ulteriori test possono aiutare a confermare la diagnosi. Questi possono includere:
Trattamento della fibrosi cistica
Poiché la fibrosi cistica è una malattia complessa, i centri di cura accreditati dalla CF Foundation adottano un approccio di squadra per gestirne il trattamento. I membri del team includono un pneumologo, un terapista della respirazione, un infermiere, un assistente sociale, un dietista e altri esperti di fibrosi cistica che vedrai regolarmente ai controlli.
Al centro di ogni team di cura della fibrosi cistica c'è la persona con FC e la sua famiglia. Sarai responsabile di una combinazione quotidiana di farmaci e altre terapie per liberare i polmoni dal muco, prevenire le infezioni e fornire il supporto nutrizionale di cui il tuo corpo ha bisogno. I trattamenti possono includere:
Farmaci per la fibrosi cistica
Il medico può prescriverti farmaci per aprire le vie aeree, fluidificare il muco, prevenire o curare infezioni e aiutare il tuo corpo a ottenere nutrienti dal cibo. Questi includono:
Antibiotici. Possono prevenire o curare le infezioni polmonari e aiutare i polmoni a funzionare meglio. Potresti assumerli sotto forma di pillole, in un inalatore o nebulizzatore o tramite un'iniezione. Potresti anche ricevere antibiotici come trattamento endovenoso, in ospedale o a casa.
Medicinali antinfiammatori. Questi includono ibuprofene e corticosteroidi come il prednisone.
Broncodilatatori. Li respirerai nei polmoni attraverso un inalatore o un nebulizzatore che trasforma il medicinale liquido in una nebbia. I broncodilatatori rilassano e aprono le vie respiratorie.
Diluenti del muco. Ti aiuteranno a eliminare la sporcizia dalle vie respiratorie fluidificando il muco e aiutandoti a tossirlo fuori dai polmoni. Li respirerai nei polmoni attraverso un inalatore o un nebulizzatore che trasforma la medicina liquida in una nebbia.
Integratori di enzimi pancreatici. Per sostituire gli enzimi digestivi bloccati dal muco denso nel pancreas, ingoierai queste capsule all'inizio di ogni pasto e della maggior parte degli spuntini. Gli enzimi ti aiuteranno a digerire il cibo e ad assorbire i nutrienti. Potrebbero anche venirti prescritti integratori multivitaminici per compensare i bassi livelli causati da problemi digestivi.
Riduttori di acidi. Le persone affette da FC spesso soffrono di reflusso acido, che si verifica quando l'acido nello stomaco ritorna nell'esofago. Pillole come gli inibitori della pompa protonica e gli anti-H2 possono ridurre il reflusso acido e aiutare gli enzimi pancreatici a funzionare meglio.
Ammorbidenti per le feci. La FC colpisce il sistema digestivo e può causare stitichezza o ristagno delle feci e portare a un'ostruzione intestinale, che può essere molto grave. Un farmaco da banco chiamato polietilenglicole (venduto come MiraLAX, GoLYTELY e altri marchi) può prevenire o trattare questi problemi.
Farmaci specifici per il diabete correlato alla fibrosi cistica (CFRD) o malattia del fegato. Il tuo team di assistenza può includere specialisti che prescriveranno e supervisioneranno i farmaci per le complicanze della FC, se necessario, come la terapia insulinica per la CFRD.
Farmaci che prendono di mira la mutazione genetica
Farmaci speciali chiamati modulatori CFTR prendono di mira il difetto sottostante nella proteina CFTR. Questi farmaci possono aiutare la proteina CFTR a funzionare correttamente, il che può rendere il muco nel corpo sottile e scivoloso. Questo può far funzionare meglio i tuoi polmoni, eliminare la tosse e aiutarti ad aumentare di peso.
I modulatori CFTR vengono assunti sotto forma di pillola, solitamente ogni 12 ore. Questi sono efficaci solo nelle persone con determinate mutazioni CFTR, inclusa F508del, che ha il 90% delle persone con FC. Attualmente sono disponibili quattro modulatori CFTR, con altri in fase di sviluppo:
Tecniche di liberazione delle vie aeree (ACT)
Questi possono aiutare a sciogliere il muco denso e appiccicoso in modo che possa essere eliminato dai polmoni tossendo o sbuffando (una tecnica che ti insegnerà il tuo terapista della respirazione). Liberare le vie aeree ogni giorno (di solito almeno due volte al giorno) può aiutare a ridurre le infezioni polmonari e a migliorare la funzionalità polmonare. Potresti provare:
Fisioterapia toracica (CPT) o percussioni. Ciò comporta picchiettare o battere le mani sul petto o sulla schiena per eliminare il muco dai polmoni. Qualcun altro lo fa per te. Assumerai posizioni diverse in modo che la gravità possa aiutare a drenare il muco dai cinque lobi dei polmoni (drenaggio posturale). Potrebbe essere necessario tossire o sbuffare per eliminare il muco sciolto dal corpo.
Oscillazione della parete toracica ad alta frequenza (il giubbotto). Si tratta di indossare un giubbotto gonfiabile collegato a una macchina. La macchina esegue la terapia fisica del torace vibrando ad alta frequenza. Il giubbotto fa vibrare il torace per sciogliere e fluidificare il muco. Durante le pause, tossirai o sbufferai per eliminare il muco.
Pressione espiratoria positiva (PEP) o PEP oscillante. Respirerai attraverso un dispositivo portatile che ti consente di inspirare normalmente ma crea resistenza quando espiri. Questo ti costringerà a espirare più forte, il che fa entrare l'aria dietro il muco nelle vie respiratorie e la fa uscire. A volte, i dispositivi provocano una vibrazione (oscillazione) per facilitare questo movimento. I marchi del dispositivo includono Flutter, Acapella e AerobikA.
Drenaggio autogeno (AD). Per fare questo, espiri forte, o sbuffa, a velocità diverse. Questo sposta il muco dalle vie aeree più piccole alle vie aeree centrali e ne facilita la fuoriuscita. Il tuo fisioterapista della FC può insegnarti la tecnica corretta.
Tecnica del ciclo attivo di respirazione (ACBT). Combina diverse tecniche di respirazione che aiutano a eliminare il muco dai polmoni in tre fasi. La prima fase ti aiuta a rilassare le vie respiratorie. La seconda fase aiuta a far passare l'aria dietro il muco e a eliminarlo. La terza fase aiuta a espellere il muco dai polmoni.
Riabilitazione polmonare
Il medico potrebbe suggerirti un programma a lungo termine per migliorare la funzionalità polmonare e la salute generale. La riabilitazione polmonare può essere eseguita in regime ambulatoriale o durante una degenza ospedaliera per un’infezione polmonare. Molte parti della riabilitazione polmonare sono incluse nelle visite cliniche regolari presso i centri di cura accreditati dalla Fondazione CF. Questi includono:
Interventi chirurgici per la fibrosi cistica
La fibrosi cistica colpisce molte parti del corpo. Il medico può raccomandare un intervento chirurgico per trattare alcune complicanze della fibrosi cistica. Come ogni intervento chirurgico, gli interventi chirurgici per la fibrosi cistica comportano il rischio di complicanze, tra cui infezioni contratte in ospedale, sanguinamento, problemi respiratori e (con gli interventi di trapianto) rigetto d'organo e infezioni. Gli interventi chirurgici possono includere:
Chirurgia nasale e dei seni. Questa procedura può rimuovere i polipi nasali (escrescenze) che ostruiscono la respirazione. La chirurgia dei seni può essere eseguita per trattare frequenti attacchi di sinusite.
Posizionamento di una sonda per l'alimentazione. Anche con l'uso di enzimi pancreatici, la fibrosi cistica interferisce con la digestione e l'assorbimento dei nutrienti dal cibo. Ciò può rendere difficile aumentare o mantenere il peso. Una sonda per l'alimentazione può aiutare a fornire nutrimento e calorie extra attraverso un integratore liquido prescritto dal team di assistenza. Il tubo può essere impiantato chirurgicamente nell'addome e non ti impedirà di mangiare per via orale.
Chirurgia intestinale. L'intervento chirurgico può aiutare a rimuovere un blocco nell'intestino. Se un segmento dell'intestino si è piegato all'interno di una sezione vicina (intussuscezione), potrebbe anche essere necessaria una riparazione chirurgica.
Trapianto di polmone. Se la tua funzionalità polmonare è gravemente compromessa, hai complicazioni polmonari potenzialmente letali o gli antibiotici hanno smesso di funzionare per le infezioni polmonari, potresti essere un candidato per un trapianto di polmone. Se hai la fibrosi cistica, entrambi i polmoni devono essere sostituiti (trapianto di doppio polmone). Non avrai la fibrosi cistica nei tuoi nuovi polmoni; tuttavia, dopo un trapianto di polmone possono verificarsi altre complicanze della fibrosi cistica, come infezioni dei seni, diabete e patologie del pancreas.
Trapianto di fegato. Per una grave malattia epatica correlata alla fibrosi cistica, come la cirrosi, può essere raccomandato un trapianto di fegato. In alcune persone, un trapianto di fegato può essere combinato con trapianti di polmone o pancreas.
Altri trattamenti per la fibrosi cistica
Le terapie non chirurgiche per la fibrosi cistica possono includere:
Ossigenoterapia. Se il livello di ossigeno nel sangue diminuisce, il medico può consigliarti di respirare ossigeno puro per prevenire la pressione alta nei polmoni (ipertensione polmonare).
Ventilazione non invasiva. Questo metodo utilizza una maschera nasale o orale per fornire pressione positiva nelle vie aeree e nei polmoni durante l'inspirazione. Di solito viene utilizzato durante il sonno, spesso in combinazione con l'ossigenoterapia. La ventilazione non invasiva può ridurre il lavoro respiratorio e favorire la pulizia delle vie aeree.
Sonda nasogastrica (NG). Una sonda NG è un tipo di sonda di alimentazione temporanea che prevede l'inserimento di un sottile , tubo flessibile nel naso, nella gola e nello stomaco. Un tubo NG è il tipo meno invasivo di tubo di alimentazione perché il suo inserimento non richiede un'incisione chirurgica. Il tubo può essere inserito ogni notte e rimosso al mattino oppure lasciato in sede per giorni.
Terapie nutrizionali per la fibrosi cistica
La fibrosi cistica colpisce il sistema digestivo in molti modi. Può rendere più difficile la crescita o l'aumento di peso, causare stitichezza o blocchi intestinali, provocare reflusso acido (bruciore di stomaco), portare a una cattiva alimentazione e altre complicazioni. Il tuo team di assistenza per la fibrosi cistica esaminerà la tua dieta, insieme a eventuali integratori o farmaci di cui potresti aver bisogno per supportare la tua salute digestiva. Oltre ad assumere questi integratori e farmaci, ti potrebbe essere chiesto di:
Segui una dieta ricca di calorie e grassi. Si stima che il fabbisogno energetico (calorico) delle persone affette da FC sia da una volta e mezzo a due volte superiore rispetto a quello di chi non soffre di FC. Poiché la fibrosi cistica rende più difficile l’assorbimento dei grassi, i medici di solito consigliano che il 40% delle calorie totali provenga dai grassi. Le persone affette da fibrosi cistica che assumono farmaci modulatori del CFTR in genere non hanno bisogno di calorie più elevate, poiché questi farmaci aiutano il gene della fibrosi cistica a funzionare correttamente. Ma devono assumere i farmaci con uno spuntino ricco di grassi affinché il farmaco venga assorbito.
Segui una dieta ricca di sale. Il sale ti aiuta a mantenere il giusto equilibrio di liquidi (acqua) nel tuo corpo. Aiuta anche i muscoli a contrarsi. Non assumere abbastanza sale può interferire con la crescita, ridurre l’appetito e causare problemi come mal di stomaco, debolezza, crampi muscolari, nausea e mal di testa. Le persone affette da FC perdono molto sale con il sudore, quindi è importante mangiare cibi più salati, soprattutto durante i periodi caldi e umidi o dopo l'attività fisica.
Complicanze della fibrosi cistica
La fibrosi cistica può causare una serie di problemi respiratori (respirazione). Oltre al declino della funzionalità polmonare, queste complicazioni includono:
Bronchiectasie. Frequenti infezioni e infiammazioni polmonari indeboliscono gradualmente le pareti delle vie aeree. Ciò può farli allargare, abbassarsi e diventare cicatrici. Questa condizione è chiamata bronchiectasie, che alla fine può portare a insufficienza respiratoria.
Emottisi. Se le bronchiectasie (danni alle vie aeree) si verificano vicino ai vasi sanguigni dei polmoni e si ha un'infezione, ciò può portare a tossire sangue (emottisi). Sebbene di solito coinvolga solo una piccola quantità di sangue, può essere pericoloso per la vita.
Pneumotorace. Se l'aria penetra nello spazio che separa i polmoni dalla parete toracica, può può causare il collasso parziale o totale di un polmone. Questo è chiamato pneumotorace e si verifica più comunemente negli adulti con fibrosi cistica. Il pneumotorace spesso dà l'impressione di una sensazione di ribollimento e può causare dolore toracico improvviso e mancanza di respiro.
Infezioni croniche. Il muco denso nei polmoni e nei seni crea un ambiente ideale per la crescita di batteri e funghi. Le persone affette da FC possono spesso avere infezioni polmonari, bronchite o polmonite. Potresti contrarre un'infezione da batteri resistenti agli antibiotici e difficili da trattare.
Esacerbazioni acute. Le persone affette da fibrosi cistica possono sperimentare un peggioramento dei sintomi respiratori, come tosse con più muco e mancanza di respiro. Questa si chiama riacutizzazione e richiede un trattamento con antibiotici, in ospedale o a casa. Perdita di peso e calo di energia sono comuni durante le riacutizzazioni.
Insufficienza respiratoria. L'insufficienza respiratoria è la causa più comune di morte per fibrosi cistica. Nel corso del tempo, la malattia può danneggiare il tessuto polmonare così gravemente da non funzionare più. La funzione polmonare peggiora gradualmente fino a quando la condizione diventa pericolosa per la vita. Se la tua funzionalità polmonare diminuisce fino a un certo livello, il tuo team di cura della FC potrebbe parlare con te della possibilità di un intervento chirurgico di trapianto di polmone, che può salvarti la vita.
I polmoni non sono gli unici parte del tuo corpo danneggiata dalla fibrosi cistica. La FC colpisce anche i seguenti organi:
Pancreas. Il muco denso causato dalla fibrosi cistica blocca i dotti del pancreas. Ciò impedisce alle proteine che scompongono il cibo, chiamate enzimi digestivi, di raggiungere l'intestino. Di conseguenza, il tuo corpo ha difficoltà a ottenere i nutrienti di cui ha bisogno. Nel corso del tempo, questo può anche portare al diabete correlato alla fibrosi cistica.
Fegato. Se i condotti che rimuovono la bile si intasano, il fegato si infiamma. Ciò può portare a gravi cicatrici chiamate cirrosi.
Intestino tenue. Poiché può essere difficile scomporre gli alimenti ad alto contenuto di acido che provengono dallo stomaco, il rivestimento dell'intestino tenue può consumarsi.
Intestino crasso. Il liquido denso nello stomaco può rendere le feci più grandi e più difficili da espellere. Ciò può portare a blocchi. In alcuni casi, l’intestino può anche iniziare a ripiegarsi su se stesso come una fisarmonica, una condizione chiamata intussuscezione. Le persone affette da FC hanno inoltre da cinque a dieci volte più probabilità di sviluppare il cancro del colon-retto rispetto alla popolazione generale.
Vescica. La tosse cronica o prolungata indebolisce i muscoli della vescica. Potresti avere incontinenza da stress con FC. Ciò significa che perdi una piccola pipì quando tossisci, starnutisci, ridi o sollevi qualcosa. Sebbene sia più comune nelle donne, anche gli uomini possono averlo.
Reni. Alcune persone affette da fibrosi cistica sviluppano calcoli renali. Questi piccoli e duri ammassi di minerali possono causare nausea, vomito e dolore. Se non li tratti, potresti contrarre un'infezione ai reni.
Organi riproduttivi. La fibrosi cistica influisce sulla fertilità negli uomini e nelle donne. La maggior parte degli uomini (98%) affetti da FC nascono senza dotti deferenti, i tubi che trasportano lo sperma nel liquido seminale. Ciò si traduce in infertilità. Le donne affette da fibrosi cistica hanno un muco cervicale molto denso, che può rendere più difficile per uno spermatozoo fecondare un ovulo. L'ovulazione irregolare dovuta a una cattiva alimentazione può anche allungare i tempi di gravidanza.
Altre parti del corpo. La fibrosi cistica può anche portare a debolezza muscolare e assottigliamento delle ossa (osteoporosi). Poiché la FC altera l'equilibrio dei minerali nel sangue, può anche causare bassa pressione sanguigna, affaticamento, battito cardiaco accelerato e una sensazione generale di debolezza.
Ulteriori screening sanitari per la fibrosi cistica
Le persone affette da FC hanno un rischio maggiore di sviluppare alcune altre malattie, inclusa la fibrosi cistica- diabete correlato (CFRD), cancro del colon-retto e osteoporosi. La diagnosi precoce è importante per trattare o gestire queste condizioni. Il tuo team di assistenza per la fibrosi cistica può consigliare screening sanitari come questi:
Test di tolleranza al glucosio orale. Il diabete correlato alla fibrosi cistica (CFRD) è una delle complicanze più comuni della fibrosi cistica negli adulti. Se hai la fibrosi cistica, probabilmente verrai sottoposto a test ogni anno per la CFRD, a partire dall'età di 10 anni, con un test di tolleranza al glucosio orale (OGTT). L'OGTT è il modo migliore per diagnosticare la CFRD e di solito viene eseguito al mattino dopo un digiuno di 8 ore. Ti verrà chiesto di bere una "bevanda di glucosio" e poi la glicemia (zucchero) verrà misurata in momenti diversi.
Colonoscopia. Il rischio di cancro del colon-retto negli adulti affetti da fibrosi cistica è da cinque a dieci volte maggiore rispetto alla popolazione generale e ancora più elevato (20 volte) per le persone affette da fibrosi cistica che ricevono un trapianto di polmone o di altro organo solido. A causa di questo rischio, si raccomanda che le persone affette da FC inizino lo screening per il cancro del colon-retto con una colonscopia all'età di 40 anni (30 anni se hanno subito un trapianto di organi solidi).
Scansione assorbimetrica a raggi X a doppia energia (DEXA). Le persone affette da fibrosi cistica sono a rischio di due comuni malattie ossee: osteoporosi e osteopenia. Queste condizioni possono rendere le tue ossa deboli e fragili. Il tuo team di assistenza per la fibrosi cistica monitorerà la tua crescita attraverso altezza e peso, seguirà il tuo sviluppo durante la pubertà e controllerà i livelli di vitamina D nel sangue. Si raccomanda che le persone affette da FC si sottopongano a una scansione DEXA entro i 18 anni e ripetano la scansione ogni 1-5 anni. Una scansione DEXA è un tipo di radiografia che controlla lo spessore delle ossa.
Takeaway
Cystic la fibrosi (CF) è una malattia genetica che colpisce i polmoni, il pancreas e altri organi. Sebbene la fibrosi cistica sia una condizione grave che necessita di cure quotidiane, esistono molti modi per trattarla e nel corso degli anni si è verificato un grande miglioramento in questi trattamenti. Le persone che soffrono di fibrosi cistica ora possono aspettarsi di vivere una vita molto più lunga rispetto a coloro che ne soffrivano in passato.
Domande frequenti sulla fibrosi cistica
Qual è l'aspettativa di vita di una persona affetta da fibrosi cistica?
Le persone affette da FC continuano a vivere una vita più lunga e più sana. Il registro dei pazienti della Cystic Fibrosis Foundation raccoglie dati di pazienti che ricevono cure per la fibrosi cistica presso i centri di cura accreditati dalla CF Foundation e hanno accettato di condividere le loro informazioni sanitarie. Sulla base dei dati del Registro 2022, si prevede che l’aspettativa di vita delle persone affette da FC nate tra il 2018 e il 2022 sarà di 56 anni. I dati del Registro 2021 mostrano che si prevede che la metà dei bambini nati nel 2021 vivrà fino a 65 anni o più. Uno studio basato su studi clinici condotti su persone affette da fibrosi cistica che assumevano il nuovo farmaco modulatore CFTR a tripla combinazione ha previsto una durata di vita di oltre 71 anni.
Le persone affette da fibrosi cistica possono avere una vita normale?
La maggior parte delle persone affette da fibrosi cistica vivono una vita quotidiana normale, con la sfida di adattarsi ai farmaci quotidiani, alla terapia per la pulizia delle vie aeree e ad altri trattamenti e farmaci. I bambini affetti da FC vanno a scuola, hanno amici, amano gli hobby e possono esercitarsi e praticare sport. Molti vanno al college, si sposano e hanno una famiglia propria.
Cosa succede se non tratti la fibrosi cistica?
Le persone affette da fibrosi cistica hanno un muco denso e appiccicoso che blocca le vie aeree nei polmoni, rendendo difficile la respirazione e facilitando il rischio di infezioni. I trattamenti per la fibrosi cistica comprendono farmaci per fluidificare il muco e combattere le infezioni e terapie per eliminare il muco dalle vie aeree. La fibrosi cistica influisce anche sulla digestione, il che rende più difficile l’assorbimento dei nutrienti dal cibo. Ci sono farmaci che aiutano in questo.
Se una persona affetta da fibrosi cistica non tratta questi sintomi, probabilmente avrà frequenti infezioni polmonari, difficoltà respiratorie e danni a lungo termine ai polmoni. Possono diventare malnutriti per mancanza di nutrienti e perdere peso, il che rende anche più difficile combattere le infezioni polmonari. Senza trattamento, la fibrosi cistica può portare a insufficienza respiratoria, blocchi intestinali, insufficienza d'organo e morte.
A che età può essere diagnosticata la fibrosi cistica?
Ogni stato degli Stati Uniti effettua test sui neonati per la fibrosi cistica. Lo screening neonatale viene effettuato durante i primi giorni di vita del bambino, utilizzando solo poche gocce di sangue prelevato dal tallone. Sebbene alla maggior parte delle persone venga diagnosticata la fibrosi cistica all'età di 2 anni, ad alcuni viene diagnosticata da adulti.
Pubblicato : 2024-08-26 09:03
Per saperne di più

- Alimenti ultra-elaborati collegati ad infarto, ictus e arresto cardiaco
- Controllo motorio oculare, prestazioni cognitive collegate dopo lieve trauma cranico
- L'inulina e l'esercizio fisico supportato dalla terapia fisica riducono il dolore nell'artrosi del ginocchio
- La morte dei genitori per overdose, omicidio e suicidio aumenta il rischio di mortalità infantile
- I funzionari esaminano le morti dopo le donazioni di plasma a Winnipeg
- Xspray Pharma ripresenta la sua richiesta alla FDA per Dasynoc
Disclaimer
È stato fatto ogni sforzo per garantire che le informazioni fornite da Drugslib.com siano accurate, aggiornate -datati e completi, ma non viene fornita alcuna garanzia in tal senso. Le informazioni sui farmaci qui contenute potrebbero essere sensibili al fattore tempo. Le informazioni su Drugslib.com sono state compilate per l'uso da parte di operatori sanitari e consumatori negli Stati Uniti e pertanto Drugslib.com non garantisce che l'uso al di fuori degli Stati Uniti sia appropriato, se non diversamente indicato. Le informazioni sui farmaci di Drugslib.com non sostengono farmaci, né diagnosticano pazienti né raccomandano terapie. Le informazioni sui farmaci di Drugslib.com sono una risorsa informativa progettata per assistere gli operatori sanitari autorizzati nella cura dei propri pazienti e/o per servire i consumatori che considerano questo servizio come un supplemento e non come un sostituto dell'esperienza, dell'abilità, della conoscenza e del giudizio dell'assistenza sanitaria professionisti.
L'assenza di un'avvertenza per un determinato farmaco o combinazione di farmaci non deve in alcun modo essere interpretata come indicazione che il farmaco o la combinazione di farmaci sia sicura, efficace o appropriata per un dato paziente. Drugslib.com non si assume alcuna responsabilità per qualsiasi aspetto dell'assistenza sanitaria amministrata con l'aiuto delle informazioni fornite da Drugslib.com. Le informazioni contenute nel presente documento non intendono coprire tutti i possibili usi, indicazioni, precauzioni, avvertenze, interazioni farmacologiche, reazioni allergiche o effetti avversi. Se hai domande sui farmaci che stai assumendo, consulta il tuo medico, infermiere o farmacista.
Parole chiave popolari
- metformin obat apa
- alahan panjang
- glimepiride obat apa
- takikardia adalah
- erau ernie
- pradiabetes
- besar88
- atrofi adalah
- kutu anjing
- trakeostomi
- mayzent pi
- enbrel auto injector not working
- enbrel interactions
- lenvima life expectancy
- leqvio pi
- what is lenvima
- lenvima pi
- empagliflozin-linagliptin
- encourage foundation for enbrel
- qulipta drug interactions