Cystische fibrose
Wat is cystische fibrose?
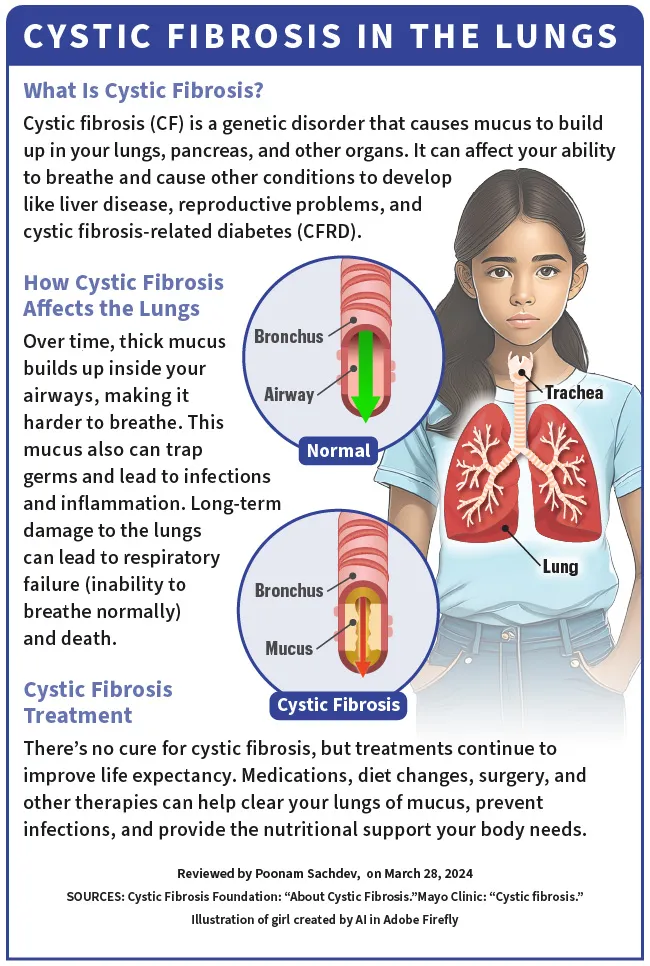
Taaislijmziekte (CF) is een genetische aandoening, wat betekent dat je deze bij de geboorte van je ouders krijgt. Het beïnvloedt uw longen, pancreas en andere organen. CF verandert de manier waarop chloride (zout) door de cellen van uw lichaam beweegt. Dit zorgt ervoor dat het slijm (dat dun en glad zou moeten zijn) in verschillende organen dik en plakkerig wordt.

Na verloop van tijd hoopt dit dikke slijm zich op in uw luchtwegen, waardoor het moeilijk wordt om te ademen . Het slijm vangt ziektekiemen op en leidt tot infecties en ontstekingen. Het kan ook ernstige, langdurige schade aan de longen veroorzaken en leiden tot ademhalingsfalen (onvermogen om normaal te ademen) en de dood.
In de alvleesklier verhindert het dikke slijm, veroorzaakt door CF, de afgifte van spijsverteringsenzymen tijdens het eten. Dit leidt tot ondervoeding en slechte groei. CF kan ook leverziekten, voortplantingsproblemen en cystische fibrose-gerelateerde diabetes (CFRD) veroorzaken.
Meer dan 40.000 mensen in de VS leven met CF. Artsen diagnosticeren elk jaar ongeveer 1.000 nieuwe gevallen. Tegenwoordig is meer dan de helft van de CF-populatie 18 jaar of ouder, en nieuwe behandelingen hebben de levensverwachting met decennia verlengd.
Cystic Fibrosis Symptomen
Mensen met CF kunnen de volgende symptomen hebben:
Atypische cystische fibrose
Er is ook een vorm van CF die 'atypische cystische fibrose' wordt genoemd. Het is een mildere vorm en kan slechts één orgaan aantasten. Symptomen verschijnen meestal veel later in het leven dan bij mensen met typische CF. In tegenstelling tot cystische fibrose bestaat er geen standaarddefinitie voor atypische cystische fibrose. Symptomen van atypische CF kunnen zijn:
Oorzaken van cystische fibrose
Taaislijmziekte wordt veroorzaakt door een verandering of mutatie in een gen genaamd CFTR (cystische fibrose transmembraangeleidingsregulator). Het eiwit in dit gen regelt de stroom van zout en vloeistoffen in en uit uw cellen. Als het CFTR-gen niet goed werkt, hoopt zich een kleverig slijm op in je lichaam.
Om CF te krijgen, moet je de gemuteerde kopie van het gen erven van je beide ouders . Er zijn meer dan 1.700 bekende mutaties van het CFTR-gen. Negentig procent van de getroffenen heeft minstens één kopie van de F508del-mutatie.
Als u slechts één exemplaar erft, heeft u geen symptomen, maar bent u wel drager van de ziekte. Dat betekent dat er een kans is dat u het aan uw kinderen kunt doorgeven.
Ongeveer 10 miljoen Amerikanen zijn drager van CF. Elke keer dat twee CF-dragers een baby krijgen, is er een kans van 25% (1 op 4) dat hun baby met CF wordt geboren.
Cystische fibrose-diagnose
Vroege diagnose betekent vroege behandeling en een betere gezondheid later in het leven. Elke staat in de VS test pasgeborenen op cystische fibrose met behulp van een of meer van deze drie tests:
Bloedtest. Deze test controleert de niveaus van immunoreactief trypsinogeen (IRT). Mensen met CF hebben hogere niveaus ervan in hun bloed. Elke staat voert ten minste één bloedtest uit voor screening op pasgeborenen.
DNA-test. Hiermee wordt gezocht naar mutaties in het CFTR-gen.
Zweettest. Deze pijnloze test meet het zout (chloride) in je zweet. Als uw resultaten hoger zijn dan normaal, duidt dit op CF.
Het diagnosticeren van CF omvat verschillende stappen. Een volledige evaluatie zou een screening van pasgeborenen, een zweetchloridetest, een genetische of dragerschapstest (DNA) en een klinische evaluatie in een erkend zorgcentrum moeten omvatten.
De meeste mensen met CF zijn gediagnosticeerd op de leeftijd van 2 jaar. Bij sommige mensen die bij de geboorte niet zijn getest, wordt de diagnose CF pas gesteld als ze volwassen zijn. Uw arts kan u DNA- of zweettesten geven als u symptomen van CF heeft.
Een zweetchloridetest is de meest betrouwbare manier om CF te diagnosticeren.
Als uw baby een bloedtest heeft ondergaan die op cystische fibrose wijst, maar een tussenliggende (niet doorslaggevende ) zweettest kan de arts bij uw baby de diagnose CFTR-gerelateerd metabool syndroom (CRMS) stellen. De vooruitzichten van iemand met CRMS zijn onduidelijk, maar ze lopen mogelijk een groter risico op problemen met de luchtwegen, sinussen, het voortplantingssysteem, de darmen of de alvleesklier.
Als een zweettest of genetische test geen uitsluitsel geeft, kunnen twee andere tests helpen bij het diagnosticeren van CF:
Nasaal potentiaalverschil (NPD). Er wordt er een kleine elektrische stroom door de neuswand (epitheel) geleid. Er worden verschillende oplossingen op de neuswand aangebracht en de elektrische stroom wordt gemeten. Mensen met CF reageren heel anders op deze test dan mensen zonder CF.
Darmstroommeting (ICM). Het omvat een pijnloze biopsie (laboratoriumtest van monsterweefsel) van rectaal weefsel om de CFTR-functie van cellen te testen.
Hoewel bloed-, DNA- en zweettesten de meest gebruikelijke en vertrouwde methoden zijn voor het diagnosticeren van CF, kunnen aanvullende tests helpen de diagnose te bevestigen. Deze kunnen het volgende omvatten:
Behandeling van cystische fibrose
Omdat cystische fibrose een complexe ziekte is, hanteren de door de CF Foundation geaccrediteerde zorgcentra een teambenadering bij het beheer van de behandeling. Tot de teamleden behoren een longarts, ademhalingstherapeut, verpleegkundige, maatschappelijk werker, diëtist en andere CF-experts die u regelmatig bij controles zult zien.
In het middelpunt van elk CF-zorgteam staat de persoon met CF en zijn/haar familie. U bent verantwoordelijk voor een dagelijkse combinatie van medicijnen en andere therapieën om slijm uit uw longen te verwijderen, infecties te voorkomen en de voedingsondersteuning te bieden die uw lichaam nodig heeft. Behandelingen kunnen het volgende omvatten:
medicijnen tegen cystische fibrose
uw arts kan u medicijnen voorschrijven om uw luchtwegen te openen, slijm te verdunnen, infecties te voorkomen of te behandelen, en help uw lichaam voedingsstoffen uit voedsel te halen. Deze omvatten:
Antibiotica. Ze kunnen longinfecties voorkomen of behandelen en ervoor zorgen dat uw longen beter werken. U kunt ze krijgen in de vorm van pillen, in een inhalator of vernevelaar, of via een injectie. U kunt ook antibiotica krijgen als IV-behandeling, in een ziekenhuis of thuis.
Ontstekingsremmende geneesmiddelen. Deze omvatten ibuprofen en corticosteroïden zoals prednison.
Bronchusverwijders. U ademt deze in uw longen via een inhalator of een vernevelaar die het vloeibare medicijn in een mist verandert. Luchtwegverwijders ontspannen en openen uw luchtwegen.
Slijmverdunners. Ze helpen je de smurrie uit je luchtwegen te krijgen door het slijm te verdunnen en je te helpen het uit je longen te hoesten. U ademt deze in uw longen via een inhalator of een vernevelaar die het vloeibare medicijn in een mist verandert.
Pancreasenzymsupplementen. Om de spijsverteringsenzymen te vervangen die worden geblokkeerd door dik slijm in de alvleesklier, slikt u deze capsules aan het begin van elke maaltijd en de meeste snacks. Enzymen helpen je je voedsel te verteren en voedingsstoffen te absorberen. Mogelijk krijgt u ook multivitaminesupplementen voorgeschreven ter compensatie van de lage concentraties veroorzaakt door spijsverteringsproblemen.
Zuurverlagende middelen. Mensen met CF hebben vaak last van zure reflux, wat betekent dat het maagzuur zich opstapelt. in de slokdarm. Pillen zoals protonpompremmers en H2-blokkers kunnen zure reflux verminderen en ervoor zorgen dat uw pancreasenzymen beter werken.
Ontlastingverzachters. CF tast het spijsverteringsstelsel aan en kan constipatie of ontlasting veroorzaken, wat kan leiden tot darmobstructie, wat zeer ernstig kan zijn. Een vrij verkrijgbaar medicijn genaamd polyethyleenglycol (verkocht als MiraLAX, GoLYTELY en andere merken) kan deze problemen voorkomen of behandelen.
Specifieke medicijnen voor cystische fibrose-gerelateerde diabetes (CFRD) of leverziekte. Uw zorgteam kan bestaan uit specialisten die, indien nodig, medicijnen voor complicaties van CF voorschrijven en daar toezicht op houden, zoals insulinetherapie voor CFRD.
Medicijnen die zich richten op de genetische mutatie
Speciale medicijnen, genaamd CFTR-modulatoren, richten zich op het onderliggende defect in het CFTR-eiwit. Deze medicijnen kunnen ervoor zorgen dat het CFTR-eiwit goed functioneert, waardoor het slijm in uw lichaam dun en glad kan worden. Hierdoor kunnen uw longen beter werken, kunt u van uw hoest afkomen en kunt u aankomen.
CFTR-modulatoren worden in pilvorm ingenomen, meestal elke 12 uur. Deze zijn alleen effectief bij mensen met bepaalde CFTR-mutaties, waaronder F508del, die 90% van de mensen met CF heeft. Momenteel zijn er vier CFTR-modulatoren beschikbaar, en er zijn er nog meer in ontwikkeling:
Luchtwegklaringstechnieken (ACT)
Deze kunnen helpen het dikke, plakkerige slijm los te maken, zodat het uit uw longen kan worden verwijderd door te hoesten of te hijgen (een techniek die uw ademhalingstherapeut u zal leren). Elke dag de luchtwegen vrijmaken (meestal minstens twee keer per dag) kan longinfecties helpen verminderen en de longfunctie verbeteren. U kunt het volgende proberen:
Borstfysiotherapie (CPT) of percussie. Dit houdt in dat u op uw borst of rug tikt of klapt om het slijm uit uw longen te verwijderen. Iemand anders doet dit voor u. U komt in verschillende houdingen terecht, zodat de zwaartekracht kan helpen het slijm uit de vijf longkwabben af te voeren (houdingsdrainage). Mogelijk moet u hoesten of snuiven om het losgekomen slijm uit uw lichaam te verwijderen.
Hoogfrequente borstwandoscillatie (het vest). Hierbij wordt een opblaasbaar vest gedragen dat aan een machine is bevestigd. Het apparaat voert borstfysiotherapie uit door met een hoge frequentie te trillen. Het vest trilt op de borst om het slijm los te maken en te verdunnen. Tijdens pauzes hoest of snoof je om het slijm te verwijderen.
Positieve uitademingsdruk (PEP) of oscillerende PEP. Je ademt door een draagbaar apparaat waarmee je normaal kunt inademen, maar weerstand creëert als je uitademt. Dit dwingt je om harder uit te ademen, waardoor lucht achter het slijm in je luchtwegen terechtkomt en naar buiten wordt verplaatst. Soms veroorzaken de apparaten een trilling (oscillatie) om deze beweging te vergemakkelijken. Merknamen voor het apparaat zijn onder meer Flutter, Acapella en AerobikA.
Autogene drainage (AD). Om dit te doen, adem je hard uit, of zucht, met verschillende snelheden. Hierdoor wordt het slijm van uw kleinere luchtwegen naar de centrale luchtwegen verplaatst, waardoor het gemakkelijker wordt om eruit te komen. Uw CF-fysiotherapeut kan u de juiste techniek leren.
Actieve cyclus van ademhalingstechniek (ACBT). Deze combineert verschillende ademhalingstechnieken die in drie fasen helpen slijm uit de longen te verwijderen. De eerste fase helpt u uw luchtwegen te ontspannen. De tweede fase helpt je om lucht achter het slijm te krijgen en het slijm te verwijderen. De derde fase helpt het slijm uit je longen te verwijderen.
Longrevalidatie
Uw arts kan u een langetermijnprogramma voorstellen om uw longfunctie en algehele gezondheid te verbeteren. Longrevalidatie kan poliklinisch worden uitgevoerd of tijdens een verblijf in het ziekenhuis vanwege een longinfectie. Veel onderdelen van de longrevalidatie maken deel uit van reguliere kliniekbezoeken aan door de CF Foundation geaccrediteerde zorgcentra. Deze omvatten:
Operaties voor cystische fibrose
CF treft vele delen van het lichaam. Uw arts kan een operatie aanbevelen om bepaalde complicaties van CF te behandelen. Zoals bij elke operatie brengen CF-operaties een risico met zich mee op complicaties, waaronder ziekenhuisinfecties, bloedingen, ademhalingsproblemen en (bij transplantatieoperaties) orgaanafstoting en infecties. Operaties kunnen het volgende omvatten:
Neus- en sinuschirurgie. Met deze procedure kunnen neuspoliepen (gezwellen) worden verwijderd die de ademhaling belemmeren. Er kan een sinusoperatie worden uitgevoerd om frequente aanvallen van sinusitis te behandelen.
Plaatsing van de voedingssonde. Zelfs met het gebruik van pancreasenzymen interfereert CF met de vertering en opname van voedingsstoffen uit voedsel. Dit kan het moeilijk maken om op gewicht te komen of te blijven. Een voedingssonde kan helpen extra voeding en calorieën toe te dienen via een vloeibaar supplement dat uw zorgteam voorschrijft. Het slangetje kan operatief in de buik worden geïmplanteerd en zal u er niet van weerhouden om via de mond te eten.
Darmoperatie. Een operatie kan helpen een verstopping in uw darm te verwijderen. Als een deel van de darm zich in een nabijgelegen gedeelte heeft gevouwen (invaginatie), kan er ook een chirurgische reparatie nodig zijn.
Longtransplantatie. Als uw longfunctie ernstig is achteruitgegaan, u levensbedreigende longcomplicaties heeft, of antibiotica niet meer werken bij longinfecties, komt u mogelijk in aanmerking voor een longtransplantatie. Als u CF heeft, moeten beide longen vervangen worden (dubbele longtransplantatie). U zult geen CF in uw nieuwe longen hebben; andere complicaties van CF, zoals sinusinfecties, diabetes en pancreasaandoeningen, kunnen echter nog steeds optreden na een longtransplantatie.
Levertransplantatie. Voor ernstige CF-gerelateerde leverziekte, zoals cirrose, kan een levertransplantatie worden aanbevolen. Bij sommige mensen kan een levertransplantatie gecombineerd worden met long- of pancreastransplantaties.
Andere behandelingen voor cystische fibrose
Niet-chirurgische therapieën voor CF kunnen het volgende omvatten:
Zuurstoftherapie. Als het zuurstofniveau in uw bloed daalt, kan uw arts aanbevelen dat u zuivere zuurstof inademt om hoge bloeddruk in de longen (pulmonale hypertensie) te voorkomen.
Niet-invasieve beademing. Bij deze methode wordt gebruik gemaakt van een neus- of mondmasker om positieve druk in de luchtwegen en de longen te creëren wanneer je inademt. De methode wordt meestal gebruikt tijdens het slapen, vaak in combinatie met zuurstoftherapie. Niet-invasieve beademing kan de ademhalingsarbeid verminderen en helpen bij het vrijmaken van de luchtwegen.
Nasogastrische (NG) sonde. Een NG-sonde is een soort tijdelijke voedingssonde waarbij een dunne slang wordt ingebracht. flexibele slang in uw neus, in uw keel en in uw maag. Een NG-sonde is het minst invasieve type voedingssonde, omdat voor het inbrengen ervan geen chirurgische incisie nodig is. De sonde kan elke nacht worden ingebracht en 's ochtends worden verwijderd, of kan dagenlang op zijn plaats blijven.
Voedingstherapieën voor cystische fibrose
Taaislijmziekte heeft op veel manieren invloed op het spijsverteringsstelsel. Het kan het moeilijker maken om te groeien of aan te komen, constipatie of darmblokkades veroorzaken, zure reflux (brandend maagzuur) veroorzaken, tot slechte voeding leiden en andere complicaties. Uw CF-zorgteam zal uw dieet beoordelen, samen met eventuele supplementen of medicijnen die u mogelijk nodig heeft om uw spijsvertering te ondersteunen. Naast het nemen van deze supplementen en medicijnen, kan u gevraagd worden om:
Volg een calorierijk en vetrijk dieet. De energiebehoefte (calorische behoeften) van mensen met CF is naar schatting anderhalf tot twee keer hoger dan die van mensen zonder CF. Omdat CF het moeilijker maakt om vet te absorberen, adviseren artsen gewoonlijk dat 40% van uw totale calorieën uit vet moet komen. Mensen met CF die CFTR-modulatormedicijnen gebruiken, hebben doorgaans niet de hogere calorieën nodig, omdat deze medicijnen het CF-gen helpen goed te werken. Maar ze moeten de medicijnen innemen met een vetrijk tussendoortje, zodat het medicijn wordt opgenomen.
Volg een zoutrijk dieet. Zout helpt je de juiste vochtbalans (water) in je lichaam te behouden. Het helpt ook de spieren samentrekken. Als u niet genoeg zout binnenkrijgt, kan dit de groei belemmeren, de eetlust verminderen en problemen veroorzaken zoals maagpijn, zwakte, spierkrampen, misselijkheid en hoofdpijn. Mensen met CF verliezen veel zout in hun zweet, dus het is belangrijk om meer zout voedsel te eten, vooral tijdens warm, vochtig weer of na het sporten.
Complicaties van cystische fibrose
Taaislijmziekte kan een aantal ademhalingsproblemen veroorzaken. Naast een afnemende longfunctie omvatten deze complicaties onder meer:
Bronchiëctasie. Frequente longinfecties en -ontstekingen verzwakken geleidelijk de wanden van de luchtwegen. Hierdoor kunnen ze groter worden, doorzakken en littekens krijgen. Deze aandoening wordt bronchiëctasie genoemd en kan uiteindelijk leiden tot ademhalingsfalen.
Bloedspuwing. Als bronchiëctasie (beschadiging van de luchtwegen) optreedt in de buurt van bloedvaten in de longen en u een infectie heeft, kan dit leiden tot het ophoesten van bloed (bloedspuwing). Hoewel er meestal maar een kleine hoeveelheid bloed bij betrokken is, kan het levensbedreigend zijn.
Pneumothorax. Als lucht lekt in de ruimte die de longen scheidt van de borstwand, kan dit levensbedreigend zijn. kan ertoe leiden dat een deel of de gehele long instort. Dit wordt pneumothorax genoemd en komt vaker voor bij volwassenen met CF. Een pneumothorax voelt vaak aan als een borrelend gevoel en kan plotselinge pijn op de borst en kortademigheid veroorzaken.
Chronische infecties. Dik slijm in de longen en sinussen creëert een ideale omgeving voor bacteriën en schimmels om te groeien. Mensen met CF kunnen vaak longinfecties, bronchitis of longontsteking hebben. U kunt besmet raken met bacteriën die resistent zijn tegen antibiotica en moeilijk te behandelen zijn.
Acute exacerbaties. Mensen met CF kunnen een verergering van hun ademhalingssymptomen ervaren, zoals hoesten met meer slijm en kortademigheid. Dit wordt een acute exacerbatie genoemd en vereist behandeling met antibiotica, hetzij in het ziekenhuis, hetzij thuis. Gewichtsverlies en lagere energie komen vaak voor tijdens exacerbaties.
Ademhalingsfalen. Ademhalingsfalen is de meest voorkomende doodsoorzaak door CF. Na verloop van tijd kan de ziekte het longweefsel zo ernstig beschadigen dat het niet meer werkt. De longfunctie verslechtert geleidelijk totdat de toestand levensbedreigend wordt. Als uw longfunctie tot een bepaald niveau afneemt, kan uw CF-zorgteam met u praten over de mogelijkheid van een longtransplantatie, die levensreddend kan zijn.
De longen zijn niet de enige deel van uw lichaam CF-schade. CF tast ook de volgende organen aan:
Alvleesklier. Het dikke slijm veroorzaakt door CF blokkeert de kanalen in uw alvleesklier. Dit voorkomt dat eiwitten die uw voedsel afbreken, spijsverteringsenzymen genoemd, uw darm bereiken. Als gevolg hiervan heeft uw lichaam moeite om de voedingsstoffen binnen te krijgen die het nodig heeft. Na verloop van tijd kan dit ook leiden tot diabetes die verband houdt met cystische fibrose.
Lever. Als de buizen die de gal afvoeren verstopt raken, raakt uw lever ontstoken. Dit kan leiden tot ernstige littekens, cirrose genaamd.
Dunne darm. Omdat het moeilijk kan zijn om zuurrijk voedsel dat uit je maag komt af te breken, kan het slijmvlies van de dunne darm wegslijten.
> Dikke darm. Het dikke vocht in uw maag kan uw ontlasting groot en moeilijker maken. Dit kan tot verstoppingen leiden. In sommige gevallen kan uw darm zich ook als een accordeon in zichzelf opvouwen, een aandoening die darminvaginatie wordt genoemd. Mensen met CF hebben ook vijf tot tien keer meer kans om colorectale kanker te ontwikkelen dan de algemene bevolking.
Blaas. Chronisch of langdurig hoesten verzwakt uw blaasspieren. Het kan zijn dat u stress-incontinentie heeft met CF. Dit betekent dat je een beetje urine verliest als je hoest, niest, lacht of iets optilt. Hoewel het vaker voorkomt bij vrouwen, kunnen mannen het ook hebben.
Nieren. Sommige mensen met CF krijgen nierstenen. Deze kleine, harde clusters van mineralen kunnen misselijkheid, braken en pijn veroorzaken. Als u ze niet behandelt, kunt u een nierinfectie krijgen.
Voortplantingsorganen. CF beïnvloedt de vruchtbaarheid bij mannen en vrouwen. De meeste mannen (98%) met CF worden geboren zonder zaadleider, de buisjes die sperma naar sperma transporteren. Dit resulteert in onvruchtbaarheid. Vrouwen met CF hebben zeer dik baarmoederhalsslijm, waardoor het voor een spermacel moeilijker kan worden een eicel te bevruchten. Een onregelmatige ovulatie als gevolg van slechte voeding kan er ook voor zorgen dat een zwangerschap langer duurt.
Andere delen van het lichaam. CF kan ook leiden tot spierzwakte en dunner wordende botten (osteoporose). Omdat CF de mineralenbalans in uw bloed verstoort, kan het ook een lage bloeddruk, vermoeidheid, een snelle hartslag en een algemeen gevoel van zwakte veroorzaken.
Aanvullende gezondheidsonderzoeken voor cystische fibrose
Mensen met CF hebben een hoger risico op het ontwikkelen van bepaalde andere ziekten, waaronder cystische fibrose- gerelateerde diabetes (CFRD), colorectale kanker en osteoporose. Vroege detectie is belangrijk om deze aandoeningen te behandelen of te beheersen. Uw CF-zorgteam kan gezondheidsonderzoeken aanbevelen zoals deze:
Orale glucosetolerantietest. Cystic fibrosis-gerelateerde diabetes (CFRD) is een van de meest voorkomende complicaties van CF bij volwassenen. Als u CF heeft, wordt u vanaf 10-jarige leeftijd waarschijnlijk elk jaar getest op CFRD met een orale glucosetolerantietest (OGTT). De OGTT is de beste manier om CFRD te diagnosticeren en wordt meestal 's morgens gedaan na 8 uur vasten. U wordt gevraagd een “glucosedrank” te drinken en vervolgens wordt uw bloedglucose (suiker) op verschillende tijdstippen gemeten.
Colonoscopie. Het risico op colorectale kanker bij volwassenen met cystische fibrose is vijf tot tien keer groter dan bij de algemene bevolking, en zelfs hoger (20 keer) voor mensen met CF die een long- of andere solide orgaantransplantatie ondergaan. Vanwege dit risico wordt aanbevolen dat mensen met CF op 40-jarige leeftijd beginnen met screening op colorectale kanker met een colonoscopie (op 30-jarige leeftijd als u een solide orgaantransplantatie heeft gehad).
DEXA-scan (Dual-Energy X-ray Absorptiometry). Mensen met CF lopen risico op twee veel voorkomende botziekten: osteoporose en osteopenie. Deze omstandigheden kunnen uw botten zwak en broos maken. Uw CF-zorgteam zal uw groei in lengte en gewicht volgen, uw ontwikkeling in de puberteit volgen en uw bloed controleren op vitamine D-waarden. Het wordt aanbevolen dat mensen met CF vóór hun 18e een DEXA-scan laten maken en de scan elke 1-5 jaar herhalen. Een DEXA-scan is een soort röntgenfoto die de dikte van uw botten controleert.
Takeaways
Cystic fibrose (CF) is een genetische ziekte die de longen, pancreas en andere organen aantast. Hoewel CF een ernstige aandoening is die dagelijkse zorg nodig heeft, zijn er veel manieren om het te behandelen, en er is in de loop der jaren een grote verbetering in die behandelingen opgetreden. Mensen die nu CF hebben, kunnen een veel langer leven verwachten dan degenen die het in het verleden hadden.
Veelgestelde vragen over cystische fibrose
Wat is de levensverwachting van iemand met cystische fibrose?
Mensen met CF blijven langer en gezonder leven. Het patiëntenregister van de Cystic Fibrosis Foundation verzamelt gegevens van patiënten die zorg voor CF ontvangen in door de CF Foundation geaccrediteerde zorgcentra en die hebben ingestemd met het delen van hun gezondheidsinformatie. Op basis van de gegevens uit het Register uit 2022 wordt voorspeld dat de levensverwachting van mensen met CF die tussen 2018 en 2022 geboren zijn, 56 jaar bedraagt. Gegevens uit het Register 2021 laten zien dat de helft van de baby’s die in 2021 geboren worden, naar verwachting 65 jaar of ouder zal worden. Een onderzoek gebaseerd op klinische onderzoeken bij mensen met CF die de nieuwere drievoudige combinatie CFTR-modulatormedicatie gebruikten, voorspelde een mogelijke levensduur van meer dan 71 jaar.
Kunnen mensen met cystische fibrose een normaal leven leiden?
De meeste mensen met CF leiden een normaal dagelijks leven, met de uitdaging van het inpassen van dagelijkse medicijnen, luchtwegklaringstherapie en andere behandelingen en medicijnen. Kinderen met CF gaan naar school, hebben vrienden, hebben hobby’s en kunnen sporten en sporten. Velen gaan naar de universiteit, trouwen en hebben een eigen gezin.
Wat gebeurt er als je cystische fibrose niet behandelt?
Mensen met CF hebben dik, plakkerig slijm dat de luchtwegen blokkeert in hun longen, waardoor ze moeilijk kunnen ademen en gemakkelijker infecties kunnen krijgen. Behandelingen voor CF omvatten medicijnen om het slijm te verdunnen en infecties te bestrijden, en therapieën om het slijm uit de luchtwegen te verwijderen. CF heeft ook invloed op de spijsvertering, waardoor het moeilijker wordt om voedingsstoffen uit voedsel op te nemen. Er zijn medicijnen die hierbij helpen.
Als een persoon met CF deze symptomen niet behandelt, zal hij of zij waarschijnlijk frequente longinfecties, ademhalingsmoeilijkheden en langdurige schade aan de longen krijgen. Ze kunnen ondervoed raken door een gebrek aan voedingsstoffen en afvallen, waardoor het ook moeilijker wordt om longinfecties te bestrijden. Zonder behandeling kan CF leiden tot respiratoire insufficiëntie, darmblokkades, orgaanfalen en overlijden.
Op welke leeftijd kan bij u de diagnose cystische fibrose worden gesteld? /p> Elke staat in de VS test pasgeborenen op cystische fibrose. De screening van pasgeborenen wordt uitgevoerd tijdens de eerste paar dagen van het leven van een baby, waarbij slechts een paar druppels bloed uit de hiel worden gebruikt. Hoewel bij de meeste mensen op 2-jarige leeftijd de diagnose CF wordt gesteld, worden sommige mensen als volwassenen gediagnosticeerd. Geplaatst : 2024-08-26 09:03 Er is alles aan gedaan om ervoor te zorgen dat de informatie die wordt verstrekt door Drugslib.com accuraat en up-to-date is -datum en volledig, maar daarvoor wordt geen garantie gegeven. De hierin opgenomen geneesmiddelinformatie kan tijdgevoelig zijn. De informatie van Drugslib.com is samengesteld voor gebruik door zorgverleners en consumenten in de Verenigde Staten en daarom garandeert Drugslib.com niet dat gebruik buiten de Verenigde Staten gepast is, tenzij specifiek anders aangegeven. De geneesmiddeleninformatie van Drugslib.com onderschrijft geen geneesmiddelen, diagnosticeert geen patiënten of beveelt geen therapie aan. De geneesmiddeleninformatie van Drugslib.com is een informatiebron die is ontworpen om gelicentieerde zorgverleners te helpen bij de zorg voor hun patiënten en/of om consumenten te dienen die deze service zien als een aanvulling op en niet als vervanging voor de expertise, vaardigheden, kennis en beoordelingsvermogen van de gezondheidszorg. beoefenaars. Het ontbreken van een waarschuwing voor een bepaald medicijn of een bepaalde medicijncombinatie mag op geen enkele manier worden geïnterpreteerd als een indicatie dat het medicijn of de medicijncombinatie veilig, effectief of geschikt is voor een bepaalde patiënt. Drugslib.com aanvaardt geen enkele verantwoordelijkheid voor enig aspect van de gezondheidszorg die wordt toegediend met behulp van de informatie die Drugslib.com verstrekt. De informatie in dit document is niet bedoeld om alle mogelijke toepassingen, aanwijzingen, voorzorgsmaatregelen, waarschuwingen, geneesmiddelinteracties, allergische reacties of bijwerkingen te dekken. Als u vragen heeft over de medicijnen die u gebruikt, neem dan contact op met uw arts, verpleegkundige of apotheker.Lees verder

Disclaimer
Populaire zoekwoorden