Fibrose cística
O que é fibrose cística?
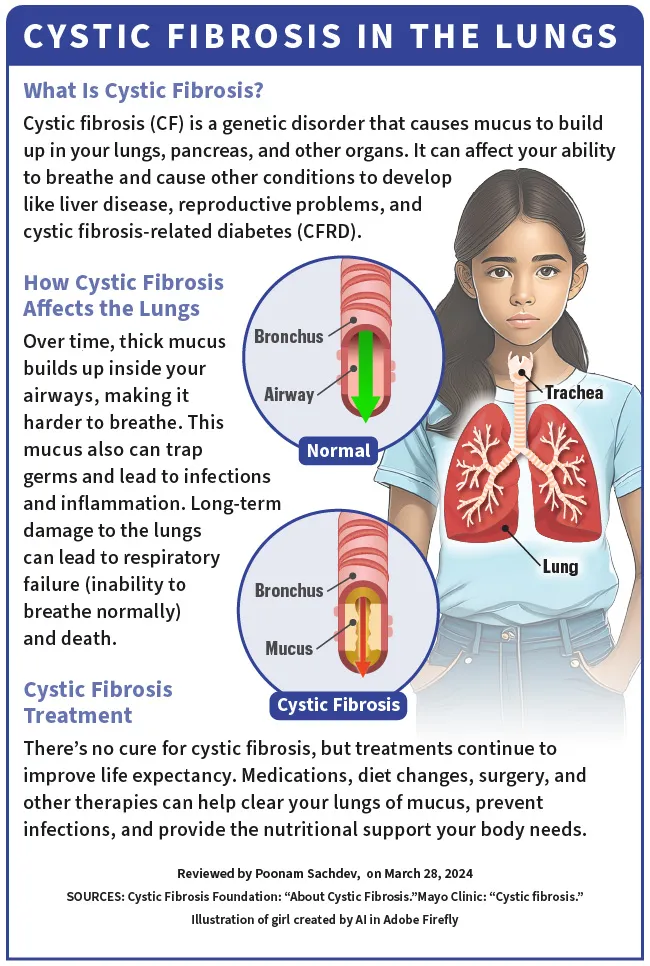
A fibrose cística (FC) é uma doença genética, o que significa que você a contrai dos seus pais ao nascer. Afeta os pulmões, o pâncreas e outros órgãos. A FC muda a forma como o cloreto (sal) se move através das células do seu corpo. Isso faz com que o muco (que deveria ser fino e escorregadio) em vários órgãos se torne espesso e pegajoso.
Com o tempo, esse muco espesso se acumula dentro das vias respiratórias, dificultando a respiração. . O muco retém germes e causa infecções e inflamações. Também pode causar danos graves e de longo prazo aos pulmões e levar à insuficiência respiratória (incapacidade de respirar normalmente) e à morte.
No pâncreas, o muco espesso causado pela FC impede a liberação de enzimas digestivas quando você come. Isto leva à desnutrição e ao fraco crescimento. A FC também pode causar doenças hepáticas, problemas reprodutivos e diabetes relacionado à fibrose cística (DRFC).
Mais de 40.000 pessoas nos EUA vivem com FC. Os médicos diagnosticam cerca de 1.000 novos casos a cada ano. Hoje, mais de metade da população com FC tem 18 anos ou mais e novos tratamentos aumentaram a esperança de vida em décadas.
Sintomas de fibrose cística
Pessoas com FC podem ter sintomas que incluem:
Fibrose cística atípica
Existe também uma forma de FC chamada “fibrose cística atípica”. É um tipo mais brando e pode afetar apenas um órgão. Os sintomas geralmente aparecem muito mais tarde na vida do que em pessoas com FC típica. Ao contrário da fibrose cística, não existe uma definição padrão para fibrose cística atípica. Os sintomas da FC atípica podem incluir:
Causas da fibrose cística
A fibrose cística é causada por uma alteração, ou mutação, em um gene chamado CFTR (regulador de condutância transmembrana da fibrose cística). A proteína deste gene controla o fluxo de sal e fluidos para dentro e para fora das células. Se o gene CFTR não funcionar corretamente, um muco pegajoso se acumula em seu corpo.
Para contrair FC, você precisa herdar a cópia mutada do gene de ambos os seus pais. . Existem mais de 1.700 mutações conhecidas do gene CFTR. Noventa por cento das pessoas afetadas têm pelo menos uma cópia da mutação F508del.
Se você herdar apenas uma cópia, não terá nenhum sintoma, mas será portador da doença. Isso significa que há uma chance de você transmiti-lo aos seus filhos.
Cerca de 10 milhões de americanos são portadores de FC. Cada vez que dois portadores de FC têm um bebê, há 25% (1 em 4) de chance de seu bebê nascer com FC.
Diagnóstico de Fibrose Cística
O diagnóstico precoce significa tratamento precoce e melhor saúde mais tarde na vida. Cada estado dos EUA testa recém-nascidos para fibrose cística usando um ou mais destes três testes:
Exame de sangue. Este teste verifica os níveis de tripsinogênio imunorreativo (IRT). Pessoas com FC apresentam níveis mais elevados no sangue. Cada estado realiza pelo menos um exame de sangue para triagem neonatal.
Teste de DNA. Isso procura mutações no gene CFTR.
Teste do suor. Este teste indolor mede o sal (cloreto) no suor. Se seus resultados forem superiores ao normal, isso sugere FC.
O diagnóstico de FC envolve várias etapas. Uma avaliação completa deve incluir uma triagem neonatal, um teste de cloreto no suor, um teste genético ou de portador (DNA) e uma avaliação clínica em um centro de atendimento credenciado.
A maioria das pessoas com FC tem diagnosticado aos 2 anos de idade. Algumas pessoas que não foram testadas ao nascer não são diagnosticadas com FC até se tornarem adultas. Seu médico poderá fazer testes de DNA ou de suor se você tiver sintomas de FC.
Um teste de cloreto no suor é a maneira mais confiável de diagnosticar FC.
Se o seu bebê fizer um exame de sangue que indique fibrose cística, mas tiver um resultado intermediário (inconclusivo) ) teste do suor, o médico pode diagnosticar seu bebê com síndrome metabólica relacionada à CFTR (CRMS). A perspectiva de uma pessoa com CRMS não é clara, mas ela pode ter um risco maior de problemas nas vias aéreas, seios da face, sistema reprodutivo, intestinos ou pâncreas.
Se um teste do suor ou um teste genético for inconclusivo, dois outros testes podem ajudar a diagnosticar a FC:
Diferença de potencial nasal (NPD). Isso envolve a passagem de uma pequena corrente elétrica pelo revestimento nasal (epitélio). Diferentes soluções são aplicadas no revestimento nasal e a corrente elétrica é medida. Pessoas com FC respondem a este teste de maneira muito diferente daquelas sem FC.
Medição da corrente intestinal (ICM). Envolve uma biópsia indolor (teste laboratorial de amostra de tecido) do tecido retal para testar a função CFTR das células.
Embora os testes de sangue, DNA e suor sejam os métodos mais comuns e confiáveis para diagnosticar FC, testes adicionais podem ajudar a confirmar o diagnóstico. Estes podem incluir:
Tratamento de Fibrose Cística
Como a fibrose cística é uma doença complexa, os centros de atendimento credenciados pela CF Foundation adotam uma abordagem de equipe para gerenciar seu tratamento. Os membros da equipe incluem um pneumologista, terapeuta respiratório, enfermeiro, assistente social, nutricionista e outros especialistas em FC que você verá regularmente em exames.
No centro de toda equipe de tratamento de FC está a pessoa com FC e sua família. Você será responsável por uma combinação diária de medicamentos e outras terapias para limpar o muco dos pulmões, prevenir infecções e fornecer o suporte nutricional de que seu corpo necessita. Os tratamentos podem incluir:
Medicamentos para fibrose cística
Seu médico pode prescrever medicamentos para abrir as vias respiratórias, diluir o muco, prevenir ou tratar infecções e ajudar seu corpo a obter nutrientes dos alimentos. Isso inclui:
Antibióticos. Eles podem prevenir ou tratar infecções pulmonares e ajudar os pulmões a funcionar melhor. Você pode obtê-los na forma de comprimidos, em um inalador ou nebulizador ou por meio de uma injeção. Você também pode receber antibióticos como tratamento intravenoso, no hospital ou em casa.
Medicamentos antiinflamatórios. Isso inclui ibuprofeno e corticosteróides como a prednisona.
Broncodilatadores. Você os inalará nos pulmões por meio de um inalador ou nebulizador que transforma o medicamento líquido em uma névoa. Os broncodilatadores relaxam e abrem as vias respiratórias.
Diluidores de muco. Eles ajudarão você a tirar a gosma das vias respiratórias, diluindo o muco e ajudando você a expulsá-lo dos pulmões. Você os respirará para os pulmões por meio de um inalador ou nebulizador que transforma o medicamento líquido em uma névoa.
Suplementos de enzimas pancreáticas. Para substituir as enzimas digestivas que são bloqueadas pelo muco espesso no pâncreas, você engolirá essas cápsulas no início de cada refeição e na maioria dos lanches. As enzimas o ajudarão a digerir os alimentos e a absorver os nutrientes. Também podem ser prescritos suplementos multivitamínicos para compensar os níveis baixos causados por problemas digestivos.
Redutores de ácido. Pessoas com FC geralmente apresentam refluxo ácido, que ocorre quando o ácido estomacal aumenta. para o esôfago. Pílulas como inibidores da bomba de prótons e bloqueadores H2 podem reduzir o refluxo ácido e ajudar as enzimas pancreáticas a funcionar melhor.
Amaciadores de fezes. A FC afeta o sistema digestivo e pode causar prisão de ventre ou acúmulo de fezes e levar à obstrução intestinal, que pode ser muito grave. Um medicamento de venda livre chamado polietilenoglicol (vendido como MiraLAX, GoLYTELY e outras marcas) pode prevenir ou tratar esses problemas.
Medicamentos específicos para diabetes relacionado à fibrose cística (DRFC) ou doença hepática. Sua equipe médica pode incluir especialistas que prescreverão e supervisionarão medicamentos para complicações da FC conforme necessário, como terapia com insulina para CFRD.
Medicamentos que têm como alvo a mutação genética
Medicamentos especiais chamados moduladores de CFTR têm como alvo o defeito subjacente na proteína CFTR. Esses medicamentos podem ajudar a proteína CFTR a funcionar corretamente, o que pode tornar o muco do corpo fino e escorregadio. Isso pode fazer com que seus pulmões funcionem melhor, eliminar a tosse e ajudar você a ganhar peso.
Moduladores CFTR são tomados em forma de pílula, geralmente a cada 12 horas. Estes são eficazes apenas em pessoas com certas mutações CFTR, incluindo F508del, que 90% das pessoas com FC têm. Atualmente, existem quatro moduladores CFTR disponíveis, com mais em desenvolvimento:
Técnicas de desobstrução das vias aéreas (ACT)
Isso pode ajudar a soltar o muco espesso e pegajoso, para que ele possa ser eliminado dos pulmões ao tossir ou bufar (uma técnica que seu terapeuta respiratório lhe ensinará). A limpeza das vias aéreas todos os dias (geralmente pelo menos duas vezes ao dia) pode ajudar a reduzir infecções pulmonares e melhorar a função pulmonar. Você pode tentar:
Fisioterapia torácica (CPT) ou percussão. Isso envolve bater palmas no peito ou nas costas para limpar o muco dos pulmões. Outra pessoa faz isso por você. Você ficará em posições diferentes para que a gravidade possa ajudar a drenar o muco dos cinco lóbulos dos pulmões (drenagem postural). Pode ser necessário tossir ou bufar para limpar o muco solto do corpo.
Oscilação da parede torácica de alta frequência (o colete). Isso envolve o uso de um colete inflável preso a uma máquina. A máquina realiza fisioterapia torácica vibrando em alta frequência. O colete vibra no peito para soltar e diluir o muco. Durante as pausas, você tossirá ou bufará para limpar o muco.
Pressão expiratória positiva (PEP) ou PEP oscilante. Você respirará por meio de um dispositivo portátil que permite inspirar normalmente, mas cria resistência ao expirar. Isso forçará você a expirar com mais força, o que deixa o ar atrás do muco nas vias respiratórias e o expulsa. Às vezes, os aparelhos causam uma vibração (oscilação) para ajudar nesse movimento. As marcas do dispositivo incluem Flutter, Acapella e AerobikA.
Drenagem autogênica (DA). Para fazer isso, você expira com força ou bufa em velocidades diferentes. Isso move o muco das vias aéreas menores para as vias aéreas centrais e facilita sua saída. Seu fisioterapeuta com FC pode lhe ensinar a técnica adequada.
Técnica de ciclo ativo de respiração (TCCA). Isso combina diferentes técnicas de respiração que ajudam a limpar o muco dos pulmões em três fases. A primeira fase ajuda a relaxar as vias respiratórias. A segunda fase ajuda a colocar ar atrás do muco e a limpá-lo. A terceira fase ajuda a expulsar o muco dos pulmões.
Reabilitação pulmonar
Seu médico pode sugerir um programa de longo prazo para melhorar sua função pulmonar e sua saúde geral. A reabilitação pulmonar pode ser feita em regime ambulatorial ou durante a internação hospitalar devido a uma infecção pulmonar. Muitas partes da reabilitação pulmonar estão incluídas em visitas clínicas regulares em centros de atendimento credenciados pela CF Foundation. Estes incluem:
Cirurgias para fibrose cística
A FC afeta muitas partes do corpo. O seu médico pode recomendar cirurgia para tratar certas complicações da FC. Como qualquer cirurgia, as cirurgias de FC apresentam risco de complicações, incluindo infecções adquiridas no hospital, sangramento, problemas respiratórios e (com cirurgias de transplante) rejeição de órgãos e infecções. As cirurgias podem incluir:
Cirurgia nasal e sinusal. Este procedimento pode remover pólipos nasais (crescimentos) que obstruem a respiração. A cirurgia sinusal pode ser realizada para tratar crises frequentes de sinusite.
Colocação de sonda de alimentação. Mesmo com o uso de enzimas pancreáticas, a FC interfere na digestão e absorção de nutrientes dos alimentos. Isso pode dificultar o ganho ou a manutenção do peso. Um tubo de alimentação pode ajudar a fornecer nutrição e calorias extras por meio de um suplemento líquido prescrito pela equipe médica. O tubo pode ser implantado cirurgicamente no abdômen e não impedirá que você coma pela boca.
Cirurgia intestinal. A cirurgia pode ajudar a remover uma obstrução no intestino. Se um segmento do intestino se dobrar dentro de uma seção próxima (intussuscepção), também pode ser necessário um reparo cirúrgico.
Transplante de pulmão. Se a sua função pulmonar diminuiu gravemente, você tem complicações pulmonares potencialmente fatais ou os antibióticos pararam de funcionar para infecções pulmonares, você pode ser um candidato a um transplante de pulmão. Se você tem FC, ambos os pulmões precisam ser substituídos (transplante duplo de pulmão). Você não terá FC em seus novos pulmões; no entanto, outras complicações da FC, como infecções sinusais, diabetes e doenças do pâncreas, ainda podem ocorrer após um transplante de pulmão.
Transplante de fígado. Para doença hepática grave relacionada à FC, como cirrose, um transplante de fígado pode ser recomendado. Em algumas pessoas, um transplante de fígado pode ser combinado com transplantes de pulmão ou pâncreas.
Outros tratamentos para fibrose cística
As terapias não cirúrgicas para FC podem incluir:
Oxigenoterapia. Se o seu nível de oxigênio no sangue diminuir, seu médico poderá recomendar que você respire oxigênio puro para evitar pressão alta nos pulmões (hipertensão pulmonar).
Ventilação não invasiva. Este método usa uma máscara nasal ou bucal para fornecer pressão positiva nas vias aéreas e nos pulmões quando você inspira. Geralmente é usado durante o sono, geralmente em combinação com oxigenoterapia. A ventilação não invasiva pode diminuir o trabalho respiratório e ajudar na desobstrução das vias aéreas.
Sonda nasogástrica (NG). Uma sonda NG é um tipo de opção de sonda de alimentação temporária que envolve a inserção de um tubo fino , tubo flexível no nariz, na garganta e no estômago. Uma sonda NG é o tipo menos invasivo de sonda de alimentação porque sua inserção não requer uma incisão cirúrgica. O tubo pode ser inserido todas as noites e removido pela manhã ou deixado no local por dias.
Terapias nutricionais para fibrose cística
A fibrose cística afeta o sistema digestivo de várias maneiras. Pode dificultar o crescimento ou ganho de peso, causar prisão de ventre ou bloqueios intestinais, causar refluxo ácido (azia), levar à má nutrição e outras complicações. Sua equipe de tratamento de FC revisará sua dieta, juntamente com quaisquer suplementos ou medicamentos necessários para apoiar sua saúde digestiva. Além de tomar esses suplementos e medicamentos, você pode ser solicitado a:
Faça uma dieta rica em calorias e gorduras. Estima-se que as necessidades energéticas (calóricas) das pessoas com FC sejam uma vez e meia a duas vezes maiores do que aquelas sem FC. Como a FC dificulta a absorção de gordura, os médicos geralmente aconselham que 40% do total de calorias sejam provenientes de gordura. Pessoas com FC que tomam medicamentos moduladores de CFTR normalmente não precisam de calorias mais altas, pois esses medicamentos ajudam o gene da FC a funcionar corretamente. Mas eles precisam tomar os medicamentos com um lanche rico em gordura para que o medicamento seja absorvido.
Faça uma dieta rica em sal. O sal ajuda a manter o equilíbrio correto de fluidos (água) em seu corpo. Também ajuda a contrair os músculos. Não ingerir sal suficiente pode interferir no crescimento, reduzir o apetite e causar problemas como dor de estômago, fraqueza, cãibras musculares, náuseas e dor de cabeça. Pessoas com FC perdem muito sal no suor, por isso é importante comer mais alimentos salgados, especialmente durante o tempo quente e úmido ou após o exercício.
Complicações da Fibrose Cística
A fibrose cística pode causar vários problemas respiratórios. Além do declínio da função pulmonar, essas complicações incluem:
Bronquiectasia. Infecções pulmonares frequentes e inflamações enfraquecem gradualmente as paredes das vias aéreas. Isso pode fazer com que eles se alarguem, caiam e fiquem com cicatrizes. Essa condição é chamada de bronquiectasia, que pode eventualmente levar à insuficiência respiratória.
Hemoptise. Se ocorrer bronquiectasia (dano nas vias aéreas) perto dos vasos sanguíneos nos pulmões e você tiver uma infecção, isso pode causar tosse com sangue (hemoptise). Embora geralmente envolva apenas uma pequena quantidade de sangue, pode ser fatal.
Pneumotórax. Se o ar vazar para o espaço que separa os pulmões da parede torácica, ele pode causar o colapso de parte ou de todo o pulmão. Isso é chamado de pneumotórax e ocorre mais comumente em adultos com FC. O pneumotórax geralmente parece uma sensação de borbulhamento e pode causar dor repentina no peito e falta de ar.
Infecções crônicas. O muco espesso nos pulmões e seios da face cria um ambiente ideal para o crescimento de bactérias e fungos. Pessoas com FC podem frequentemente ter infecções pulmonares, bronquite ou pneumonia. Você pode ser infectado por bactérias resistentes aos antibióticos e difíceis de tratar.
Exacerbações agudas. Pessoas com FC podem apresentar piora dos sintomas respiratórios, como tosse com mais muco e falta de ar. Isto é chamado de exacerbação aguda e requer tratamento com antibióticos, no hospital ou em casa. Perda de peso e menor energia são comuns durante as exacerbações.
Insuficiência respiratória. A insuficiência respiratória é a causa mais comum de morte por FC. Com o tempo, a doença pode danificar tanto o tecido pulmonar que ele não funciona mais. A função pulmonar piora gradualmente até que a condição se torne fatal. Se a sua função pulmonar diminuir até um certo nível, a sua equipa de cuidados de FC poderá falar consigo sobre a possibilidade de uma cirurgia de transplante pulmonar, que pode salvar vidas.
Os pulmões não são os únicos parte do seu corpo sofre danos de FC. A FC também afeta os seguintes órgãos:
Pâncreas. O muco espesso causado pela FC bloqueia os dutos do pâncreas. Isso impede que as proteínas que decompõem os alimentos, chamadas enzimas digestivas, cheguem ao intestino. Como resultado, seu corpo tem dificuldade em obter os nutrientes de que necessita. Com o tempo, isso também pode levar ao diabetes relacionado à fibrose cística.
Fígado. Se os tubos que removem a bile ficarem obstruídos, seu fígado ficará inflamado. Isso pode causar cicatrizes graves chamadas cirrose.
Intestino delgado. Como pode ser difícil decompor alimentos com alto teor de ácido provenientes do estômago, o revestimento do intestino delgado pode se desgastar.
Intestino grosso. O líquido espesso no estômago pode tornar as fezes grandes e mais difíceis de evacuar. Isso pode levar a bloqueios. Em alguns casos, o intestino também pode começar a se dobrar como um acordeão, uma condição chamada intussuscepção. Pessoas com FC também têm cinco a dez vezes mais probabilidade de desenvolver câncer colorretal do que a população em geral.
Bexiga. A tosse crônica ou prolongada enfraquece os músculos da bexiga. Você pode ter incontinência de esforço com FC. Isso significa que você vaza um pouco de xixi quando tosse, espirra, ri ou levanta alguma coisa. Embora seja mais comum em mulheres, os homens também podem ter.
Rins. Algumas pessoas com FC apresentam pedras nos rins. Esses pequenos e duros aglomerados de minerais podem causar náuseas, vômitos e dor. Se você não tratá-los, poderá contrair uma infecção renal.
Órgãos reprodutivos. A FC afeta a fertilidade em homens e mulheres. A maioria dos homens (98%) com FC nasce sem canais deferentes, os tubos que transportam os espermatozoides para o sêmen. Isso resulta em infertilidade. Mulheres com FC têm muco cervical muito espesso, o que pode dificultar a fertilização do óvulo pelo espermatozoide. A ovulação irregular devido à má nutrição também pode fazer com que a gravidez demore mais para ser alcançada.
Outras partes do corpo. A FC também pode causar fraqueza muscular e enfraquecimento dos ossos (osteoporose). Como a FC perturba o equilíbrio dos minerais no sangue, ela também pode causar pressão arterial baixa, fadiga, frequência cardíaca acelerada e uma sensação geral de fraqueza.
Exames de saúde adicionais para fibrose cística
Pessoas com FC têm um risco maior de desenvolver outras doenças, incluindo fibrose cística- diabetes relacionado (CFRD), câncer colorretal e osteoporose. A detecção precoce é importante para tratar ou controlar essas condições. Sua equipe de tratamento de FC pode recomendar exames de saúde como estes:
Teste oral de tolerância à glicose. O diabetes relacionado à fibrose cística (DRFC) é uma das complicações mais comuns da FC em adultos. Se você tem FC, provavelmente será testado todos os anos para CFRD, a partir dos 10 anos de idade, com um teste oral de tolerância à glicose (OGTT). O TOTG é a melhor forma de diagnosticar a DRFC e geralmente é feito pela manhã, após um jejum de 8 horas. Você será solicitado a beber uma “bebida de glicose” e então sua glicose (açúcar) no sangue será medida em momentos diferentes.
Colonoscopia. O risco de câncer colorretal em adultos com fibrose cística é cinco a dez vezes maior do que na população em geral, e ainda maior (20 vezes) para pessoas com FC que recebem um transplante de pulmão ou outro órgão sólido. Devido a esse risco, é recomendado que as pessoas com FC comecem o rastreamento do câncer colorretal com uma colonoscopia aos 40 anos (30 anos se você tiver feito um transplante de órgão sólido).
Absorciometria de raios X de dupla energia (DEXA). Pessoas com FC correm risco de contrair duas doenças ósseas comuns: osteoporose e osteopenia. Estas condições podem tornar os seus ossos fracos e quebradiços. Sua equipe de tratamento de FC acompanhará seu crescimento por meio de altura e peso, acompanhará seu desenvolvimento na puberdade e verificará seus níveis de vitamina D no sangue. É recomendado que as pessoas com FC façam um exame DEXA aos 18 anos e repitam o exame a cada 1-5 anos. Uma varredura DEXA é um tipo de raio X que verifica a espessura dos ossos.
Conclusões
Cística a fibrose (FC) é uma doença genética que afeta os pulmões, o pâncreas e outros órgãos. Embora a FC seja uma doença grave que necessita de cuidados diários, há muitas maneiras de tratá-la e tem havido uma grande melhoria nesses tratamentos ao longo dos anos. As pessoas que têm FC agora podem esperar viver uma vida muito mais longa do que aquelas que a tiveram no passado.
Perguntas frequentes sobre fibrose cística
Qual é a expectativa de vida de alguém com fibrose cística?
Pessoas com FC continuam a viver vidas mais longas e saudáveis. O Registro de Pacientes da Cystic Fibrosis Foundation coleta dados de pacientes que recebem cuidados para FC em centros de atendimento credenciados pela CF Foundation e que concordaram em compartilhar suas informações de saúde. Com base nos dados do Registro de 2022, a expectativa de vida das pessoas com FC que nasceram entre 2018 e 2022 está prevista em 56 anos. Os dados do Registro de 2021 mostram que se prevê que metade dos bebês nascidos em 2021 viverão até os 65 anos ou mais. Um estudo baseado em ensaios clínicos de pessoas com FC que tomaram o mais novo medicamento modulador CFTR de combinação tripla previu uma possível expectativa de vida de mais de 71 anos.
As pessoas com fibrose cística podem ter uma vida normal?
A maioria das pessoas com FC vive uma vida diária normal, com o desafio de adaptação em medicamentos diários, terapia de desobstrução das vias aéreas e outros tratamentos e medicamentos. As crianças com FC vão à escola, têm amigos, gostam de passatempos e podem fazer exercício e praticar desporto. Muitos vão para a faculdade, se casam e têm sua própria família.
O que acontece se você não tratar a fibrose cística?
Pessoas com FC têm muco espesso e pegajoso que bloqueia as vias aéreas nos pulmões, dificultando a respiração e facilitando a contração de infecções. Os tratamentos para a FC incluem medicamentos para diluir o muco e combater infecções, e terapias para limpar o muco das vias respiratórias. A FC também afeta a digestão, o que dificulta a absorção dos nutrientes dos alimentos. Existem medicamentos para ajudar com isso.
Se uma pessoa com FC não tratar esses sintomas, provavelmente terá infecções pulmonares frequentes, dificuldade para respirar e danos pulmonares de longo prazo. Eles podem ficar desnutridos por falta de nutrientes e perder peso, o que também dificulta o combate às infecções pulmonares. Sem tratamento, a FC pode causar insuficiência respiratória, obstruções intestinais, falência de órgãos e morte.
Com que idade você pode ser diagnosticado com fibrose cística?
Todos os estados dos EUA testam recém-nascidos para fibrose cística. A triagem neonatal é feita durante os primeiros dias de vida do bebê, utilizando apenas algumas gotas de sangue do calcanhar. Embora a maioria das pessoas seja diagnosticada com FC aos 2 anos de idade, algumas são diagnosticadas na idade adulta.
Postou : 2024-08-26 09:03
Consulte Mais informação

- FDA pode permitir alguns vaporizadores com sabor destinados a adultos
- Cerca de 3.000 Wayfair Dressers foram recolhidos devido ao risco de tombamento de crianças
- Mulheres têm maior probabilidade de sobreviver ao câncer do que homens – com um custo
- Venglustat da Sanofi recebe designação de terapia inovadora nos EUA para doença de Gaucher tipo 3
- FDA dos EUA concede aprovação total do Kite’s Tecartus para pacientes adultos com linfoma de células do manto recidivante ou refratário
- AbbVie anuncia resultados positivos de um estudo de fase 1 de múltiplas doses ascendentes de ABBV-295, um análogo de amilina de ação prolongada, em adultos
Isenção de responsabilidade
Todos os esforços foram feitos para garantir que as informações fornecidas por Drugslib.com sejam precisas, atualizadas -date e completo, mas nenhuma garantia é feita nesse sentido. As informações sobre medicamentos aqui contidas podem ser sensíveis ao tempo. As informações do Drugslib.com foram compiladas para uso por profissionais de saúde e consumidores nos Estados Unidos e, portanto, o Drugslib.com não garante que os usos fora dos Estados Unidos sejam apropriados, a menos que especificamente indicado de outra forma. As informações sobre medicamentos do Drugslib.com não endossam medicamentos, diagnosticam pacientes ou recomendam terapia. As informações sobre medicamentos do Drugslib.com são um recurso informativo projetado para ajudar os profissionais de saúde licenciados a cuidar de seus pacientes e/ou para atender os consumidores que veem este serviço como um complemento, e não um substituto, para a experiência, habilidade, conhecimento e julgamento dos cuidados de saúde. profissionais.
A ausência de uma advertência para um determinado medicamento ou combinação de medicamentos não deve de forma alguma ser interpretada como indicação de que o medicamento ou combinação de medicamentos é seguro, eficaz ou apropriado para qualquer paciente. Drugslib.com não assume qualquer responsabilidade por qualquer aspecto dos cuidados de saúde administrados com a ajuda das informações fornecidas por Drugslib.com. As informações aqui contidas não se destinam a cobrir todos os possíveis usos, instruções, precauções, advertências, interações medicamentosas, reações alérgicas ou efeitos adversos. Se você tiver dúvidas sobre os medicamentos que está tomando, consulte seu médico, enfermeiro ou farmacêutico.
Palavras -chave populares
- metformin obat apa
- alahan panjang
- glimepiride obat apa
- takikardia adalah
- erau ernie
- pradiabetes
- besar88
- atrofi adalah
- kutu anjing
- trakeostomi
- mayzent pi
- enbrel auto injector not working
- enbrel interactions
- lenvima life expectancy
- leqvio pi
- what is lenvima
- lenvima pi
- empagliflozin-linagliptin
- encourage foundation for enbrel
- qulipta drug interactions