Fibroza chistica
Ce este fibroza chistică?
Fibroza chistică (FC) este o afecțiune genetică, ceea ce înseamnă că o obțineți de la părinți la naștere. Îți afectează plămânii, pancreasul și alte organe. CF modifică modul în care clorura (sarea) se deplasează prin celulele corpului dumneavoastră. Acest lucru face ca mucusul (care ar trebui să fie subțire și alunecos) din diferite organe să devină gros și lipicios.

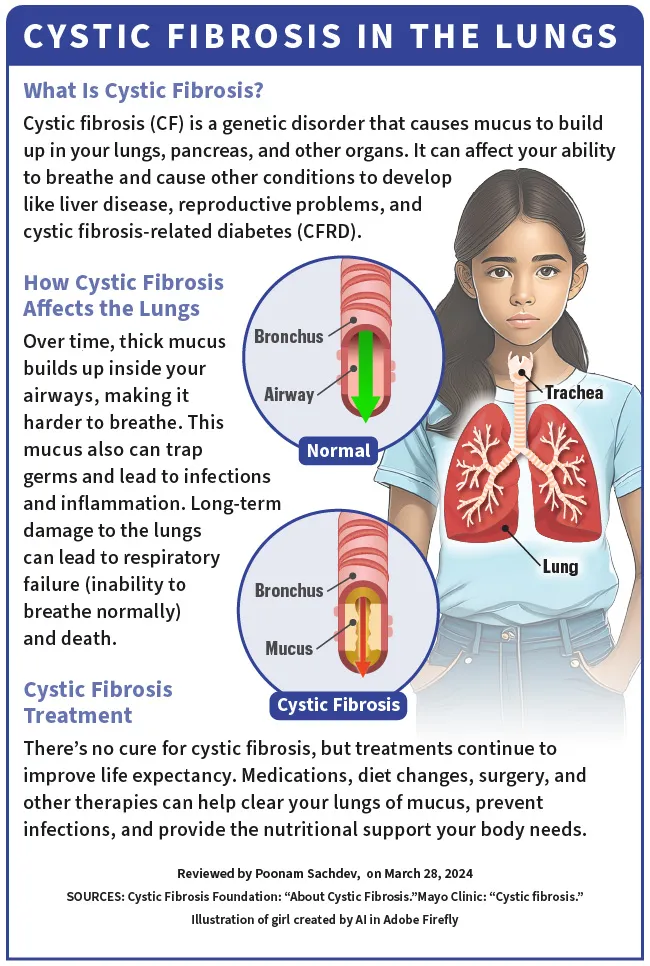
De-a lungul timpului, acest mucus gros se acumulează în interiorul căilor respiratorii, ceea ce face dificilă respirația. . Mucusul captează germenii și duce la infecții și inflamații. De asemenea, poate provoca leziuni severe și pe termen lung ale plămânilor și poate duce la insuficiență respiratorie (incapacitate de a respira normal) și moarte.
În pancreas, mucusul gros cauzat de CF împiedică eliberarea enzimelor digestive atunci când mănânci. Acest lucru duce la malnutriție și la o creștere slabă. CF poate provoca, de asemenea, boli hepatice, probleme de reproducere și diabet asociat cu fibroza chistică (CFRD).
Peste 40.000 de persoane din SUA trăiesc cu FC. Medicii diagnostichează aproximativ 1.000 de cazuri noi în fiecare an. Astăzi, mai mult de jumătate din populația cu FC are 18 ani sau mai mult, iar noile tratamente au extins speranța de viață cu zeci de ani.
Simptome de fibroză chistică
Persoanele cu FC pot avea simptome care includ:
Fibroza chistică atipică
Există și o formă de FC numită „fibroză chistică atipică”. Este un tip mai blând și poate afecta doar un organ. Simptomele apar de obicei mult mai târziu în viață decât la persoanele cu CF tipice. Spre deosebire de fibroza chistică, nu există o definiție standard pentru fibroza chistică atipică. Simptomele CF atipice pot include:
Cauzele fibrozei chistice
Fibroza chistică este cauzată de o modificare sau mutație a unei gene numită CFTR (regulator de conductanță transmembranară a fibrozei chistice). Proteina din această genă controlează fluxul de sare și fluide în și din celulele tale. Dacă gena CFTR nu funcționează corect, în corpul tău se acumulează un mucus lipicios.
Pentru a face CF, trebuie să moșteniți copia mutantă a genei de la ambii părinți. . Există peste 1.700 de mutații cunoscute ale genei CFTR. Nouăzeci la sută dintre cei afectați au cel puțin o copie a mutației F508del.
Dacă moșteniți o singură copie, nu veți avea niciun simptom, dar veți fi purtător al bolii. Asta înseamnă că există șanse să le transmiteți copiilor dvs.
Aproximativ 10 milioane de americani sunt purtători de FC. De fiecare dată când doi purtători de FC au un copil, există o șansă de 25% (1 din 4) ca copilul lor să se nască cu FC.
Diagnosticul Fibrozei Chistice
Diagnoza precoce înseamnă un tratament precoce și o sănătate mai bună mai târziu în viață. Fiecare stat din SUA testează nou-născuții pentru fibroză chistică folosind unul sau mai multe dintre aceste trei teste:
Test de sânge. Acest test verifică nivelurile de tripsinogen imunoreactiv (IRT). Persoanele cu FC au niveluri mai mari ale acesteia în sânge. Fiecare stat efectuează cel puțin un test de sânge pentru screening-ul nou-născuților.
Test ADN. Acesta caută mutații ale genei CFTR.
Test de transpirație. Acest test nedureros măsoară sarea (clorura) din transpirație. Dacă rezultatele dvs. sunt mai mari decât cele normale, sugerează CF.
Diagnosticarea CF implică mai mulți pași. O evaluare completă ar trebui să includă un screening nou-născutului, un test de clorură de transpirație, un test genetic sau de purtător (ADN) și o evaluare clinică la un centru de îngrijire acreditat.
Majoritatea persoanelor cu FC sunt diagnosticate până la vârsta de 2 ani. Unii oameni care nu au fost testați la naștere nu sunt diagnosticați cu FC până când devin adulți. Medicul dumneavoastră vă poate face teste ADN sau transpirație dacă aveți simptome de FC.
Testul de clorură de transpirație este cel mai fiabil mod de a diagnostica FC.
Dacă copilul dumneavoastră are un test de sânge care indică fibroza chistică, dar are un intermediar (neconcludent ) test de transpirație, medicul vă poate diagnostica copilul cu sindrom metabolic legat de CFTR (CRMS). Perspectiva unei persoane cu CRMS este neclară, dar poate avea un risc mai mare de probleme la nivelul căilor respiratorii, sinusurilor, sistemului reproducător, intestinelor sau pancreasului.
Dacă un test de transpirație sau un test genetic nu este concludent, alte două teste pot ajuta la diagnosticarea FC:
Diferența de potențial nazal (NPD). Acesta implică trecerea unui mic curent electric pe mucoasa nazală (epiteliu). Pe mucoasa nazală se aplică diferite soluții și se măsoară curentul electric. Persoanele cu FC răspund la acest test foarte diferit decât cele fără FC.
Măsurarea curentului intestinal (ICM). Implica o biopsie nedureroasă (test de laborator al țesutului eșantion) a țesutului rectal pentru a testa funcția CFTR a celulelor.
Test de sânge pentru funcția pancreatică
Tratamentul fibrozei chistice
Deoarece fibroza chistică este o boală complexă, centrele de îngrijire acreditate de CF Foundation adoptă o abordare de echipă pentru gestionarea tratamentului acesteia. Membrii echipei includ un pneumolog, un terapeut respirator, o asistentă medicală, un asistent social, un dietetician și alți experți în FC pe care îi veți vedea în mod regulat la controale.
În centrul fiecărei echipe de îngrijire a FC se află persoana cu FC și familia acesteia. Veți fi responsabil pentru o combinație zilnică de medicamente și alte terapii pentru a vă curăța plămânii de mucus, pentru a preveni infecțiile și pentru a oferi suportul nutrițional de care corpul dumneavoastră are nevoie. Tratamentele pot include:
Medicamente pentru fibroza chistică
Medicul dumneavoastră vă poate administra medicamente pentru a vă deschide căile respiratorii, a subțire mucusul, a preveni sau a trata infecțiile și ajuta organismul sa obtina nutrienti din alimente. Acestea includ:
Antibiotice. Ele pot preveni sau trata infecțiile pulmonare și vă pot ajuta plămânii să funcționeze mai bine. Le puteți obține sub formă de pastile, într-un inhalator sau nebulizator sau prin injectare. De asemenea, puteți primi antibiotice ca tratament IV, fie într-un spital, fie acasă.
Medicamente antiinflamatoare. Acestea includ ibuprofenul și corticosteroizii precum prednisonul.
Bronhodilatatoare. Acestea le veți respira în plămâni printr-un inhalator sau un nebulizator care transformă medicamentul lichid într-o ceață. Bronhodilatatoarele relaxează și vă deschid căile respiratorii.
Diluează mucusul. Vă vor ajuta să eliminați murdăria din căile respiratorii prin subțierea mucusului și ajutându-vă să-l eliminați din plămâni. Acestea le vei respira în plămâni printr-un inhalator sau un nebulizator care transformă medicamentul lichid într-o ceață.
Suplimente cu enzime pancreatice. Pentru a înlocui enzimele digestive care sunt blocate de mucusul gros din pancreas, veți înghiți aceste capsule la începutul fiecărei mese și a majorității gustărilor. Enzimele vă vor ajuta să vă digerați alimentele și să absorbiți nutrienții. De asemenea, vi se pot prescrie suplimente de multivitamine pentru a compensa nivelurile scăzute cauzate de problemele digestive.
Reductoare de aciditate. Persoanele cu FC au adesea reflux de acid, care este atunci când acidul din stomac este susținut. în esofag. Pastilele precum inhibitorii pompei de protoni și blocanții H2 pot reduce refluxul acid și pot ajuta enzimele tale pancreatice să funcționeze mai bine.
Balzi de scaun. CF afectează sistemul digestiv și poate cauza constipația sau scaunul să devină susținut și să ducă la o obstrucție intestinală, care poate fi foarte gravă. Un medicament fără prescripție medicală numit polietilenglicol (comercializat sub numele de MiraLAX, GoLYTELY și alte mărci) poate preveni sau trata aceste probleme.
Medicamente specifice pentru diabetul zaharat asociat fibrozei chistice (CFRD) sau boli hepatice. Echipa dvs. de îngrijire poate include specialiști care vor prescrie și vor supraveghea medicamente pentru complicațiile CF, după cum este necesar, cum ar fi terapia cu insulină pentru CFRD.
Medicamente care vizează mutația genetică
Medicamentele speciale numite modulatori CFTR vizează defectul de bază al proteinei CFTR. Aceste medicamente pot ajuta proteina CFTR să funcționeze corect, ceea ce poate face ca mucusul din organism să fie subțire și alunecos. Acest lucru vă poate face plămânii să funcționeze mai bine, vă poate scăpa de tuse și vă poate ajuta să vă îngrășați.
Modulatorii CFTR se iau sub formă de pastile, de obicei la fiecare 12 ore. Acestea sunt eficiente doar la persoanele cu anumite mutații CFTR, inclusiv F508del, pe care le au 90% dintre persoanele cu FC. În prezent, există patru modulatoare CFTR disponibile, cu mai multe în dezvoltare:
Tehnici de curățare a căilor aeriene (ACT)
Acestea pot ajuta la slăbirea mucusului gros și lipicios, astfel încât să poată fi curățat din plămâni prin tuse sau bufnitură (o tehnică pe care o va învăța terapeutul respirator). Curățarea căilor respiratorii în fiecare zi (de obicei de cel puțin două ori pe zi) poate ajuta la reducerea infecțiilor pulmonare și la îmbunătățirea funcției pulmonare. Puteți încerca:
Fizioterapie toracică (CPT) sau percuție. Acest lucru implică lovirea sau baterea din palme pe piept sau pe spate pentru a elimina mucusul din plămâni. Altcineva face asta pentru tine. Veți ajunge în poziții diferite, astfel încât gravitația poate ajuta la drenarea mucusului din cei cinci lobi ai plămânilor (drenaj postural). S-ar putea să fie nevoie să tușiți sau să bufați pentru a elimina mucusul slăbit din corp.
Oscilatie de înaltă frecvență a peretelui toracic (Vestia). Acest lucru implică purtarea unei veste gonflabile care este atașată la o mașină. Aparatul efectuează kinetoterapie toracică prin vibrare la o frecvență înaltă. Vesta vibrează pieptul pentru a slăbi și subțire mucusul. În timpul pauzelor, veți tusi sau veți bufni pentru a elimina mucusul.
Presiune expiratorie pozitivă (PEP) sau PEP oscilantă. Veți respira printr-un dispozitiv portabil care vă permite să inspirați în mod normal, dar care creează rezistență atunci când expirați. Acest lucru vă va forța să expirați mai greu, ceea ce aduce aer în spatele mucusului din căile respiratorii și îl mută afară. Uneori, dispozitivele provoacă o vibrație (oscilație) pentru a ajuta la această mișcare. Numele de marcă pentru dispozitiv includ Flutter, Acapella și AerobikA.
Drenaj autogen (AD). Pentru a face acest lucru, expirați greu sau bufați cu viteze diferite. Acest lucru mută mucusul de la căile respiratorii mai mici către căile respiratorii centrale și face mai ușor să ieși. Kinetoterapeutul dumneavoastră CF vă poate învăța tehnica adecvată.
Ciclul activ al tehnicii de respirație (ACBT). Acesta combină diferite tehnici de respirație care ajută la curățarea mucusului din plămâni în trei faze. Prima fază vă ajută să vă relaxați căile respiratorii. A doua fază vă ajută să treceți aer în spatele mucusului și să curățați mucusul. A treia fază ajută la forțarea mucusului din plămâni.
Reabilitare pulmonară
Medicul dumneavoastră vă poate sugera un program pe termen lung pentru a vă îmbunătăți funcția pulmonară și sănătatea generală. Reabilitarea pulmonară se poate face în ambulatoriu sau în timpul unei spitalizări pentru o infecție pulmonară. Multe părți ale reabilitării pulmonare sunt incluse în vizitele regulate la clinici la centrele de îngrijire acreditate de Fundația CF. Acestea includ:
Intervenții chirurgicale pentru fibroza chistică
CF afectează multe părți ale corpului. Medicul dumneavoastră vă poate recomanda o intervenție chirurgicală pentru a trata anumite complicații ale FC. Ca orice intervenție chirurgicală, operațiile cu FC prezintă un risc de complicații, inclusiv infecții dobândite în spital, sângerări, probleme respiratorii și (în cazul operațiilor de transplant) respingere și infecții de organe. Intervențiile chirurgicale pot include:
Chirurgie nazală și sinusală. Această procedură poate elimina polipii nazali (creșteri) care obstrucționează respirația. Chirurgia sinusurilor poate fi efectuată pentru a trata crizele frecvente de sinuzită.
Plasarea tubului de alimentare. Chiar și cu utilizarea enzimelor pancreatice, FC interferează cu digestia și absorbția nutrienților din alimente. Acest lucru poate face dificilă creșterea sau menținerea în greutate. Un tub de hrănire poate ajuta la furnizarea de nutriție și calorii suplimentare printr-un supliment lichid pe care echipa dumneavoastră de îngrijire îl prescrie. Tubul poate fi implantat chirurgical în abdomen și nu vă va împiedica să mâncați pe gură.
Chirurgie intestinală. Intervenția chirurgicală poate ajuta la îndepărtarea unui blocaj din intestin. Dacă un segment al intestinului s-a pliat în interiorul unei secțiuni din apropiere (invaginație), poate necesita și o reparație chirurgicală.
Transplant pulmonar. Dacă funcția pulmonară a scăzut serios, aveți complicații pulmonare care vă pun viața în pericol sau antibioticele au încetat să funcționeze pentru infecțiile pulmonare, este posibil să fiți candidat pentru un transplant pulmonar. Dacă aveți FC, ambii plămâni trebuie înlocuiți (transplant dublu pulmonar). Nu vei avea FC în noii tăi plămâni; cu toate acestea, alte complicații ale FC, cum ar fi infecțiile sinusurilor, diabetul și afecțiunile pancreasului, pot apărea în continuare după un transplant pulmonar.
Transplant hepatic. Pentru bolile hepatice severe legate de CF, cum ar fi ciroza, se poate recomanda un transplant de ficat. La unele persoane, un transplant de ficat poate fi combinat cu transplant de plămân sau pancreas.
Alte tratamente pentru fibroza chistică
Terapiile nechirurgicale pentru FC pot include:
Oxigenoterapia. Dacă nivelul de oxigen din sânge scade, medicul dumneavoastră vă poate recomanda să respirați oxigen pur pentru a preveni hipertensiunea arterială în plămâni (hipertensiune pulmonară).
Ventilație neinvazivă. Această metodă folosește o mască de nas sau de gură pentru a furniza o presiune pozitivă în căile respiratorii și plămâni atunci când inspirați. De obicei, este utilizată în timpul somnului, adesea în combinație cu terapia cu oxigen. Ventilația neinvazivă poate reduce munca de respirație și poate ajuta la curățarea căilor respiratorii.
Tub nazogastric (NG). Un tub NG este un tip de opțiune temporară de tub de alimentare care implică inserarea unui tub subțire. , tub flexibil în nas, în gât și în stomac. Un tub NG este cel mai puțin invaziv tip de tub de alimentare, deoarece introducerea acestuia nu necesită o incizie chirurgicală. Tubul poate fi introdus în fiecare noapte și îndepărtat dimineața sau lăsat pe loc zile întregi.
Terapii nutriționale pentru fibroza chistică
Fibroza chistică afectează sistemul digestiv în multe feluri. Poate îngreuna creșterea sau creșterea în greutate, poate provoca constipație sau blocaje intestinale, poate provoca reflux acid (arsuri la stomac), poate duce la o alimentație proastă și alte complicații. Echipa dumneavoastră de îngrijire a FC vă va revizui dieta, împreună cu orice suplimente sau medicamente de care aveți nevoie pentru a vă susține sănătatea digestivă. Pe lângă administrarea acestor suplimente și medicamente, vi se poate cere să:
Mănâncă o dietă bogată în calorii și grăsimi. Nevoile de energie (calorice) ale persoanelor cu FC sunt estimate a fi de o dată și jumătate până la două ori mai mari decât cele fără FC. Deoarece CF îngreunează absorbția grăsimilor, medicii recomandă de obicei ca 40% din caloriile totale să provină din grăsimi. Persoanele cu FC care iau medicamente modulatoare CFTR, de obicei, nu au nevoie de calorii mai mari, deoarece aceste medicamente ajută gena CF să funcționeze corect. Dar trebuie să ia medicamentele cu o gustare bogată în grăsimi pentru ca medicamentul să fie absorbit.
Mănâncă o dietă bogată în sare. Sarea vă ajută să mențineți echilibrul corect de lichid (apă) din organism. De asemenea, ajută la contractarea mușchilor. Lipsa de sare poate interfera cu creșterea, poate reduce pofta de mâncare și poate cauza probleme precum dureri de stomac, slăbiciune, crampe musculare, greață și dureri de cap. Persoanele cu FC pierd multă sare în transpirație, așa că este important să mănânce mai multe alimente sărate, mai ales pe vreme caldă și umedă sau după exerciții fizice.
Complicații ale fibrozei chistice
Fibroza chistică poate provoca o serie de probleme respiratorii (respirație). Pe lângă scăderea funcției pulmonare, aceste complicații includ:
Bronșiectazie. Infecțiile și inflamațiile pulmonare frecvente slăbesc treptat pereții căilor respiratorii. Acest lucru le poate determina să se lărgească, să se lase și să devină cicatrici. Această afecțiune se numește bronșiectazie, care poate duce în cele din urmă la insuficiență respiratorie.
Hemoptizie. Dacă bronșiectazia (lezarea căilor respiratorii) apare în apropierea vaselor de sânge din plămâni și aveți o infecție, aceasta poate duce la tuse cu sânge (hemoptizie). Deși de obicei implică doar o cantitate mică de sânge, poate pune viața în pericol.
Pneumotorax. Dacă aerul se scurge în spațiul care separă plămânii de peretele toracic, acesta poate provoca colapsul unei părți sau a întregului plămân. Acesta se numește pneumotorax și apare mai frecvent la adulții cu FC. Pneumotoraxul se simte adesea ca o senzație de barbotare și poate provoca durere bruscă în piept și lipsă de aer.
Infecții cronice. Mucusul gros în plămâni și sinusuri creează un mediu ideal pentru creșterea bacteriilor și ciupercilor. Persoanele cu FC pot avea adesea infecții pulmonare, bronșită sau pneumonie. Puteți să vă infectați cu bacterii rezistente la antibiotice și greu de tratat.
Exacerbări acute. Persoanele cu FC pot prezenta o agravare a simptomelor respiratorii, cum ar fi tusea cu mai mult mucus. și dificultăți de respirație. Aceasta se numește exacerbare acută și necesită tratament cu antibiotice, fie în spital, fie acasă. Pierderea în greutate și scăderea energiei sunt frecvente în timpul exacerbărilor.
Insuficiența respiratorie. Insuficiența respiratorie este cea mai frecventă cauză de deces din cauza FC. În timp, boala poate afecta atât de grav țesutul pulmonar încât nu mai funcționează. Funcția pulmonară se înrăutățește treptat până când afecțiunea devine amenințătoare pentru viață. Dacă funcția pulmonară scade la un anumit nivel, echipa de îngrijire a FC poate vorbi cu dvs. despre posibilitatea unei operații de transplant pulmonar, care poate salva vieți.
Plămânii nu sunt singurii. o parte a corpului dumneavoastră leziuni CF. CF afectează și următoarele organe:
Pancreasul. Mucusul gros cauzat de CF blochează canalele din pancreas. Acest lucru oprește proteinele care descompun alimentele, numite enzime digestive, să ajungă în intestin. Ca urmare, organismul tău are greu să obțină nutrienții de care are nevoie. În timp, acest lucru poate duce și la diabet asociat fibrozei chistice.
Ficat. Dacă tuburile care îndepărtează bila se înfundă, ficatul tău se inflama. Acest lucru poate duce la cicatrici severe numite ciroză.
Intestinul subțire. Deoarece poate fi greu să descompune alimentele bogate în acizi care provin din stomac, mucoasa intestinului subțire se poate uza.
> Intestinul gros. Lichidul gros din stomac poate face scaunul mare și mai greu de eliminat. Acest lucru poate duce la blocaje. În unele cazuri, intestinul poate începe să se plieze pe el însuși, ca un acordeon, o afecțiune numită invaginație. Persoanele cu FC au, de asemenea, de cinci până la zece ori mai multe șanse de a dezvolta cancer colorectal decât populația generală.
Vezica urinară. Tusea cronică sau de lungă durată slăbește mușchii vezicii urinare. Este posibil să aveți incontinență de stres cu CF. Aceasta înseamnă că scurgeți puțin pipi atunci când tușiți, strănutați, râdeți sau ridicați ceva. Deși este mai frecventă la femei, o pot avea și bărbații.
Rinichi. Unii oameni cu FC suferă de pietre la rinichi. Aceste grupuri mici și dure de minerale pot provoca greață, vărsături și durere. Dacă nu le tratezi, poți să faci o infecție la rinichi.
Organe de reproducere. CF afectează fertilitatea la bărbați și femei. Majoritatea bărbaților (98%) cu FC se nasc fără canalele deferente, tuburile care transportă spermatozoizii în sperma. Acest lucru duce la infertilitate. Femeile cu FC au mucus cervical foarte gros, ceea ce poate îngreuna fertilizarea unui ovul spermatozoid. Ovulația neregulată din cauza alimentației proaste poate face ca sarcina să dureze mai mult pentru a se realiza.
Alte părți ale corpului. CF poate duce, de asemenea, la slăbiciune musculară și subțierea oaselor (osteoporoză). Deoarece CF perturbă echilibrul mineralelor din sânge, poate provoca, de asemenea, tensiune arterială scăzută, oboseală, o frecvență cardiacă accelerată și o senzație generală de slăbiciune.
Evaluări suplimentare de sănătate pentru fibroza chistică
Persoanele cu FC au un risc mai mare de a dezvolta anumite alte boli, inclusiv fibroza chistică- diabet asociat (CFRD), cancer colorectal și osteoporoză. Detectarea precoce este importantă pentru a trata sau gestiona aceste afecțiuni. Echipa dumneavoastră de îngrijire a FC vă poate recomanda analize de sănătate precum acestea:
Test de toleranță orală la glucoză. Diabetul zaharat legat de fibroza chistică (CFRD) este una dintre cele mai frecvente complicații ale FC la adulți. Dacă aveți FC, probabil că veți fi testat în fiecare an pentru CFRD, începând cu vârsta de 10 ani, cu un test oral de toleranță la glucoză (OGTT). OGTT este cel mai bun mod de a diagnostica CFRD și se face de obicei dimineața, după un post de 8 ore. Vi se va cere să beți o „băutură cu glucoză” și apoi glicemia (zahărul) vă va fi măsurată la momente diferite.
Colonoscopie. Riscul de cancer colorectal la adulții cu fibroză chistică este de cinci până la zece ori mai mare decât populația generală și chiar mai mare (de 20 de ori) pentru persoanele cu FC care primesc un transplant pulmonar sau alt organ solid. Din cauza acestui risc, se recomandă ca persoanele cu FC să înceapă depistarea cancerului colorectal cu o colonoscopie la vârsta de 40 de ani (vârsta de 30 de ani dacă ați avut un transplant de organ solid).
Scanare cu absorbție cu raze X cu energie duală (DEXA). Persoanele cu FC sunt expuse riscului de a avea două boli osoase comune: osteoporoza și osteopenia. Aceste condiții vă pot face oasele slabe și fragile. Echipa dumneavoastră de îngrijire a FC vă va urmări creșterea prin înălțime și greutate, vă va urmări dezvoltarea în perioada pubertății și vă va verifica nivelul de vitamina D din sânge. Se recomandă ca persoanele cu FC să facă o scanare DEXA până la vârsta de 18 ani și să repete scanarea la fiecare 1-5 ani. O scanare DEXA este un tip de radiografie care verifică grosimea oaselor tale.
Takeaways
Chistic fibroza (FC) este o boală genetică care afectează plămânii, pancreasul și alte organe. Deși CF este o afecțiune severă care necesită îngrijire zilnică, există multe modalități de a o trata și a existat o îmbunătățire mare a acestor tratamente de-a lungul anilor. Oamenii care au FC acum se pot aștepta să trăiască o viață mult mai lungă decât cei care au avut-o în trecut.
Întrebări frecvente despre fibroza chistică
Care este speranța de viață a unei persoane cu fibroză chistică?
Persoanele cu FC continuă să trăiască o viață mai lungă și mai sănătoasă. Registrul Pacienților Fundației pentru Fibroză Chistică colectează date de la pacienții care primesc îngrijire pentru FC la centrele de îngrijire acreditate de Fundația CF și au fost de acord să-și partajeze informațiile de sănătate. Pe baza datelor din Registrul din 2022, speranța de viață a persoanelor cu FC care s-au născut între 2018 și 2022 este estimată a fi de 56 de ani. Datele din Registrul 2021 arată că jumătate dintre copiii născuți în 2021 se estimează că vor trăi până la vârsta de 65 de ani sau mai mult. Un studiu bazat pe studii clinice asupra persoanelor cu FC care iau noul medicament modulator CFTR cu triplă combinație a prezis posibile durate de viață de peste 71 de ani.
Pot persoanele cu fibroză chistică să aibă o viață normală?
Majoritatea persoanelor cu FC duc o viață normală de zi cu zi, cu provocarea de adaptare în medicamentele zilnice, terapia de curățare a căilor respiratorii și alte tratamente și medicamente. Copiii cu FC merg la școală, au prieteni, se bucură de hobby-uri și pot face mișcare și sport. Mulți merg la facultate, se căsătoresc și au familii proprii.
Ce se întâmplă dacă nu tratezi fibroza chistică?
Persoanele cu FC au mucus gros și lipicios care blochează căile respiratorii în plămâni, făcându-le dificil să respire și să facă mai ușor infecții. Tratamentele pentru FC includ medicamente pentru subțierea mucusului și combaterea infecțiilor și terapii pentru curățarea mucusului din căile respiratorii. CF afectează și digestia, ceea ce face mai dificilă absorbția nutrienților din alimente. Există medicamente care ajută la acest lucru.
Dacă o persoană cu FC nu tratează aceste simptome, va avea probabil infecții pulmonare frecvente, dificultăți de respirație și leziuni pe termen lung ale plămânilor. Ei pot deveni subnutriți din cauza lipsei de nutrienți și pot pierde în greutate, ceea ce face, de asemenea, mai dificilă lupta împotriva infecțiilor pulmonare. Fără tratament, FC poate duce la insuficiență respiratorie, blocaje intestinale, insuficiență de organ și deces.
La ce vârstă poți fi diagnosticat cu fibroză chistică?
Fiecare stat din SUA testează nou-născuții pentru fibroză chistică. Screeningul nou-născutului se face în primele zile de viață ale bebelușului, folosind doar câteva picături de sânge din călcâi. Deși majoritatea oamenilor sunt diagnosticați cu FC până la vârsta de 2 ani, unii sunt diagnosticați ca adulți.
Postat : 2024-08-26 09:03
Citeşte mai mult

- Aldeyra Therapeutics primește scrisoare de răspuns completă de la Administrația SUA pentru Alimente și Medicamente pentru cererea de medicament nou Reproxalap pentru tratamentul semnelor și simptomelor bolii de ochi uscat
- Sănătatea orală precară a copilăriei este legată de BCV aterosclerotică la vârsta adultă
- Este posibil să nu fi fost numărate până la 155.000 de decese cauzate de COVID, potrivit unui studiu
- Cireșele dulci întunecate pot ajuta la cancerul de sân agresiv lent, sugerează un studiu pe șoarece
- FDA aprobă Hernexeos, prima terapie țintită pentru adulți cu NSCLC avansat cu mutant HER2 ca opțiune inițială de tratament
- Judecătorul federal întrerupe modificările lui Kennedy la programul de vaccinuri pentru copii
Declinare de responsabilitate
S-au depus toate eforturile pentru a se asigura că informațiile furnizate de Drugslib.com sunt exacte, actualizate -data și completă, dar nu se face nicio garanție în acest sens. Informațiile despre medicamente conținute aici pot fi sensibile la timp. Informațiile Drugslib.com au fost compilate pentru a fi utilizate de către practicienii din domeniul sănătății și consumatorii din Statele Unite și, prin urmare, Drugslib.com nu garantează că utilizările în afara Statelor Unite sunt adecvate, cu excepția cazului în care se indică altfel. Informațiile despre medicamente de la Drugslib.com nu susțin medicamente, nu diagnostichează pacienții și nu recomandă terapie. Informațiile despre medicamente de la Drugslib.com sunt o resursă informațională concepută pentru a ajuta practicienii autorizați din domeniul sănătății în îngrijirea pacienților lor și/sau pentru a servi consumatorilor care văd acest serviciu ca un supliment și nu un substitut pentru expertiza, abilitățile, cunoștințele și raționamentul asistenței medicale. practicieni.
Lipsa unui avertisment pentru un anumit medicament sau combinație de medicamente nu trebuie în niciun fel interpretată ca indicând faptul că medicamentul sau combinația de medicamente este sigură, eficientă sau adecvată pentru un anumit pacient. Drugslib.com nu își asumă nicio responsabilitate pentru niciun aspect al asistenței medicale administrat cu ajutorul informațiilor furnizate de Drugslib.com. Informațiile conținute aici nu sunt destinate să acopere toate utilizările posibile, instrucțiunile, precauțiile, avertismentele, interacțiunile medicamentoase, reacțiile alergice sau efectele adverse. Dacă aveți întrebări despre medicamentele pe care le luați, consultați medicul, asistenta sau farmacistul.
Cuvinte cheie populare
- metformin obat apa
- alahan panjang
- glimepiride obat apa
- takikardia adalah
- erau ernie
- pradiabetes
- besar88
- atrofi adalah
- kutu anjing
- trakeostomi
- mayzent pi
- enbrel auto injector not working
- enbrel interactions
- lenvima life expectancy
- leqvio pi
- what is lenvima
- lenvima pi
- empagliflozin-linagliptin
- encourage foundation for enbrel
- qulipta drug interactions