Cystic Fibrosis
What Is Cystic Fibrosis?
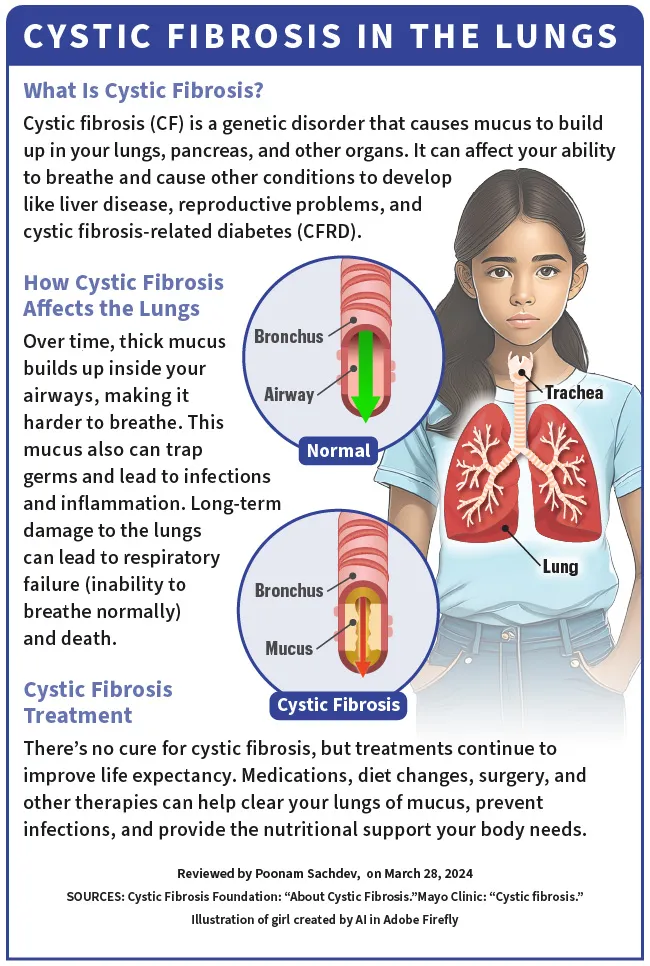
Cystic fibrosis (CF) is a genetic disorder, which means you get it from your parents at birth. It affects your lungs, pancreas, and other organs. CF changes the way chloride (salt) moves through the cells of your body. This causes the mucus (which should be thin and slippery) in various organs to become thick and sticky.
Over time, this thick mucus builds up inside your airways, making it hard to breathe. The mucus traps germs and leads to infections and inflammation. It can also cause severe, long-term damage to the lungs and lead to respiratory failure (inability to breathe normally) and death.
In the pancreas, the thick mucus caused by CF prevents the release of digestive enzymes when you eat. This leads to malnutrition and poor growth. CF can also cause liver disease, reproductive problems, and cystic fibrosis-related diabetes (CFRD).
More than 40,000 people in the U.S. live with CF. Doctors diagnose about 1,000 new cases each year. Today, more than half of the CF population is aged 18 or older, and new treatments have expanded the life expectancy by decades.
Cystic Fibrosis Symptoms
People with CF can have symptoms that include:
Atypical cystic fibrosis
There's also a form of CF called “atypical cystic fibrosis.” It's a milder type and may only affect one organ. Symptoms usually show up much later in life than in people with typical CF. Unlike cystic fibrosis, there's no standard definition for atypical cystic fibrosis. Symptoms of atypical CF may include:
Cystic Fibrosis Causes
Cystic fibrosis is caused by a change, or mutation, in a gene called CFTR (cystic fibrosis transmembrane conductance regulator). The protein in this gene controls the flow of salt and fluids in and out of your cells. If the CFTR gene doesn’t work properly, a sticky mucus builds up in your body.
To get CF, you have to inherit the mutated copy of the gene from both of your parents. There are more than 1,700 known mutations of the CFTR gene. Ninety percent of those affected have at least one copy of the F508del mutation.
If you inherit only one copy, you won’t have any symptoms, but you will be a carrier of the disease. That means there’s a chance you could pass it on to your children.
About 10 million Americans are CF carriers. Every time two CF carriers have a baby, there’s a 25% (1 in 4) chance that their baby will be born with CF.
Cystic Fibrosis Diagnosis
Early diagnosis means early treatment and better health later in life. Every state in the U.S. tests newborns for cystic fibrosis using one or more of these three tests:
Blood test. This test checks the levels of immunoreactive trypsinogen (IRT). People with CF have higher levels of it in their blood. Every state performs at least one blood test for newborn screening.
DNA test. This looks for mutations to the CFTR gene.
Sweat test. This painless test measures the salt (chloride) in your sweat. If your results are higher than normal, it suggests CF.
Diagnosing CF involves several steps. A complete evaluation should include a newborn screening, a sweat chloride test, a genetic or carrier (DNA) test, and a clinical evaluation at an accredited care center.
Most people with CF are diagnosed by age 2. Some people who weren’t tested at birth aren’t diagnosed with CF until they become adults. Your doctor might give you DNA or sweat tests if you have symptoms of CF.
A sweat chloride test is the most reliable way to diagnose CF.
If your baby has a blood test that indicates cystic fibrosis but has an intermediate (inconclusive) sweat test, the doctor may diagnose your baby with CFTR-related metabolic syndrome (CRMS). The outlook of a person with CRMS is unclear, but they may have a higher risk of problems in the airways, sinuses, reproductive system, intestines, or pancreas.
If a sweat test or genetic test is inconclusive, two other tests can help diagnose CF:
Nasal potential difference (NPD). It involves running a small electrical current across the nasal lining (epithelium). Different solutions are applied to the nasal lining and the electrical current is measured. People with CF respond to this test very differently than those without CF.
Intestinal current measurement (ICM). It involves a painless biopsy (lab test of sample tissue) of rectal tissue to test the CFTR function of cells.
While blood, DNA, and sweat tests are the most common and trusted methods for diagnosing CF, additional tests can help confirm the diagnosis. These may include:
Cystic Fibrosis Treatment
Because cystic fibrosis is a complex disease, CF Foundation-accredited care centers take a team approach to managing its treatment. Team members include a pulmonologist, respiratory therapist, nurse, social worker, dietitian, and other CF experts that you’ll see regularly at checkups.
At the center of every CF care team is the person with CF and their family. You’ll be responsible for a daily combination of medications and other therapies to clear your lungs of mucus, prevent infections, and provide the nutritional support your body needs. Treatments may include:
Cystic fibrosis medications
Your doctor may give you drugs to open your airways, thin mucus, prevent or treat infections, and help your body get nutrients from food. These include:
Antibiotics. They can prevent or treat lung infections and help your lungs work better. You might get them as pills, in an inhaler or nebulizer, or through a shot. You may also get antibiotics as an IV treatment, either in a hospital or at home.
Anti-inflammatory medicines. These include ibuprofen and corticosteroids such as prednisone.
Bronchodilators. You’ll breathe these into your lungs through an inhaler or a nebulizer that turns the liquid medicine into a mist. Bronchodilators relax and open your airways.
Mucus thinners. They’ll help you get the gunk out of your airways by thinning mucus and helping you cough it out of your lungs. You’ll breathe these into your lungs through an inhaler or a nebulizer that turns the liquid medicine into a mist.
Pancreatic enzyme supplements. To replace the digestive enzymes that are blocked by thick mucus in the pancreas, you’ll swallow these capsules at the start of every meal and most snacks. Enzymes will help you digest your food and absorb nutrients. You may also be prescribed multivitamin supplements to make up for low levels caused by digestive problems.
Acid reducers. People with CF often have acid reflux, which is when stomach acid backs up into the esophagus. Pills such as proton pump inhibitors and H2 blockers can reduce acid reflux and help your pancreatic enzymes work better.
Stool softeners. CF affects the digestive system and can cause constipation or stool to become backed up and lead to a bowel obstruction, which can be very serious. An over-the-counter medication called polyethylene glycol (sold as MiraLAX, GoLYTELY, and other brands) can prevent or treat these problems.
Specific drugs for cystic fibrosis-related diabetes (CFRD) or liver disease. Your care team may include specialists who will prescribe and oversee medication for complications of CF as needed, such as insulin therapy for CFRD.
Medications that target the genetic mutation
Special medications called CFTR modulators target the underlying defect in the CFTR protein. These drugs can help the CFTR protein to function properly, which can make the mucus in your body thin and slippery. This can make your lungs work better, get rid of your cough, and help you gain weight.
CFTR modulators are taken in pill form, usually every 12 hours. These are effective only in people with certain CFTR mutations, including F508del, which 90% of people with CF have. Currently, there are four CFTR modulators available, with more in development:
Airway clearance techniques (ACT)
These can help loosen the thick, sticky mucus so it can be cleared from your lungs by coughing or huffing (a technique your respiratory therapist will teach you). Clearing the airways every day (usually at least twice a day) can help reduce lung infections and improve lung function. You might try:
Chest physical therapy (CPT) or percussion. This involves tapping or clapping on your chest or back to clear mucus from your lungs. Someone else does this for you. You will get into different positions so gravity can help drain the mucus from the five lobes of your lungs (postural drainage). You may need to cough or huff to clear the loosened mucus from your body.
High-frequency chest wall oscillation (the Vest). This involves wearing an inflatable vest that is attached to a machine. The machine performs chest physical therapy by vibrating at a high frequency. The vest vibrates the chest to loosen and thin mucus. During pauses, you will cough or huff to clear the mucus.
Positive expiratory pressure (PEP) or oscillating PEP. You will breathe through a handheld device that allows you to inhale normally but creates resistance when you exhale. This will force you to breathe out harder, which gets air behind the mucus in your airways and moves it out. Sometimes, the devices cause a vibration (oscillation) to help with this movement. Brand names for the device include Flutter, Acapella, and AerobikA.
Autogenic drainage (AD). To do this, you breathe out hard, or huff, at different speeds. This moves mucus from your smaller airways to the central airways and makes it easier to get out. Your CF physical therapist can teach you the proper technique.
Active cycle of breathing technique (ACBT). This combines different breathing techniques that help clear mucus from the lungs in three phases. The first phase helps you relax your airways. The second phase helps you to get air behind mucus and clears mucus. The third phase helps force the mucus out of your lungs.
Pulmonary rehabilitation
Your doctor may suggest a long-term program to improve your lung function and overall health. Pulmonary rehabilitation may be done on an outpatient basis or during a hospital stay for a lung infection. Many parts of pulmonary rehabilitation are included in regular clinic visits at CF Foundation-accredited care centers. These include:
Surgeries for cystic fibrosis
CF affects many parts of the body. Your doctor may recommend surgery to treat certain complications of CF. Like any surgery, CF surgeries carry a risk for complications, including hospital-acquired infections, bleeding, respiratory problems, and (with transplant surgeries) organ rejection and infections. Surgeries may include:
Nasal and sinus surgery. This procedure can remove nasal polyps (growths) that obstruct breathing. Sinus surgery may be done to treat frequent bouts of sinusitis.
Feeding tube placement. Even with the use of pancreatic enzymes, CF interferes with digestion and absorption of nutrients from food. This can make it difficult to gain or maintain weight. A feeding tube can help deliver extra nutrition and calories through a liquid supplement your care team prescribes. The tube can be surgically implanted in the abdomen and won't stop you from eating by mouth.
Bowel surgery. Surgery can help remove a blockage in your bowel. If a segment of the intestine has folded itself inside a nearby section (intussusception), it may also require a surgical repair.
Lung transplant. If your lung function has seriously declined, you have life-threatening lung complications, or antibiotics have stopped working for lung infections, you may be a candidate for a lung transplant. If you have CF, both lungs need to be replaced (double-lung transplant). You will not have CF in your new lungs; however, other complications of CF, such as sinus infections, diabetes, and pancreas conditions, can still occur after a lung transplant.
Liver transplant. For severe CF-related liver disease, such as cirrhosis, a liver transplant may be recommended. In some people, a liver transplant may be combined with lung or pancreas transplants.
Other treatments for cystic fibrosis
Nonsurgical therapies for CF may include:
Oxygen therapy. If your blood oxygen level declines, your doctor may recommend that you breathe pure oxygen to prevent high blood pressure in the lungs (pulmonary hypertension).
Noninvasive ventilation. This method uses a nose or mouth mask to provide positive pressure in the airway and lungs when you breathe in. It's usually used while sleeping, often in combination with oxygen therapy. Noninvasive ventilation can decrease the work of breathing and help with airway clearance.
Nasogastric (NG) tube. An NG tube is a type of temporary feeding tube option that involves inserting a thin, flexible tube into your nose, down your throat, and into your stomach. An NG tube is the least invasive type of feeding tube because inserting it doesn't require a surgical incision. The tube can be inserted each night and removed in the morning, or left in place for days.
Nutritional therapies for cystic fibrosis
Cystic fibrosis affects the digestive system in many ways. It can make it harder to grow or gain weight, cause constipation or bowel blockages, give you acid reflux (heartburn), lead to poor nutrition, and other complications. Your CF care team will review your diet, along with any supplements or medications you may need to support your digestive health. In addition to taking these supplements and medications, you may be asked to:
Eat a high-calorie, high-fat diet. The energy (caloric) needs of people with CF are estimated to be one and a half to two times higher than those without CF. Since CF makes it harder for fat to be absorbed, doctors usually advise that 40% of your total calories should come from fat. People with CF who take CFTR modulator drugs typically do not need the higher calories, as these drugs help the CF gene work properly. But they need to take the drugs with a high-fat snack for the drug to be absorbed.
Eat a high-salt diet. Salt helps you maintain the right balance of fluid (water) in your body. It also helps muscles contract. Not getting enough salt can interfere with growth, reduce appetite, and cause issues such as stomach pain, weakness, muscle cramps, nausea, and headache. People with CF lose a lot of salt in their sweat, so it’s important to eat more salty foods, especially during hot, humid weather or after exercising.
Cystic Fibrosis Complications
Cystic fibrosis can cause a number of respiratory (breathing) problems. In addition to declining lung function, these complications include:
Bronchiectasis. Frequent lung infections and inflammation gradually weaken the walls of the airways. This can cause them to widen, sag, and become scarred. This condition is called bronchiectasis, which can eventually lead to respiratory failure.
Hemoptysis. If bronchiectasis (airway damage) occurs near blood vessels in the lungs and you have an infection, it can lead to coughing up blood (hemoptysis). Though it usually involves only a small amount of blood, it can be life-threatening.
Pneumothorax. If air leaks into the space that separates the lungs from the chest wall, it can cause part or all of a lung to collapse. This is called pneumothorax and occurs more commonly in adults with CF. Pneumothorax often feels like a bubbling sensation and can cause sudden chest pain and breathlessness.
Chronic infections. Thick mucus in the lungs and sinuses creates an ideal environment for bacteria and fungi to grow. People with CF may often have lung infections, bronchitis, or pneumonia. You may become infected with bacteria that are resistant to antibiotics and difficult to treat.
Acute exacerbations. People with CF may experience worsening of their respiratory symptoms, such as coughing with more mucus and shortness of breath. This is called an acute exacerbation and requires treatment with antibiotics, either in the hospital or at home. Weight loss and lower energy are common during exacerbations.
Respiratory failure. Respiratory failure is the most common cause of death from CF. Over time, the disease can damage lung tissue so badly that it no longer works. Lung function gradually worsens until the condition becomes life-threatening. If your lung function declines to a certain level, your CF care team may speak with you about the possibility of lung transplantation surgery, which can be lifesaving.
The lungs aren’t the only part of your body CF damages. CF also affects the following organs:
Pancreas. The thick mucus caused by CF blocks ducts in your pancreas. This stops proteins that break down your food, called digestive enzymes, from reaching your intestine. As a result, your body has a hard time getting the nutrients it needs. Over time, this can also lead to cystic fibrosis-related diabetes.
Liver. If the tubes that remove bile get clogged, your liver gets inflamed. This can lead to severe scarring called cirrhosis.
Small intestine. Because it can be hard to break down high-acid foods that come from your stomach, the lining of the small intestine can wear away.
Large intestine. The thick fluid in your stomach can make your stool large and harder to pass. This can lead to blockages. In some cases, your intestine may also start to fold in on itself like an accordion, a condition called intussusception. People with CF are also five to ten times more likely to develop colorectal cancer than the general population.
Bladder. Chronic or long-lasting coughing weakens your bladder muscles. You may have stress incontinence with CF. This means that you leak a little pee when you cough, sneeze, laugh, or lift something. Though it’s more common in women, men can have it, too.
Kidneys. Some people with CF get kidney stones. These small, hard clusters of minerals can cause nausea, vomiting, and pain. If you don’t treat them, you could get a kidney infection.
Reproductive organs. CF affects fertility in men and women. Most men (98%) with CF are born without vas deferens, the tubes that transport sperm into semen. This results in infertility. Women with CF have very thick cervical mucus, which can make it harder for a sperm to fertilize an egg. Irregular ovulation due to poor nutrition can also make pregnancy take longer to achieve.
Other parts of the body. CF can also lead to muscle weakness and thinning bones (osteoporosis). Because CF upsets the balance of minerals in your blood, it can also cause low blood pressure, fatigue, a fast heart rate, and a general feeling of weakness.
Additional health screenings for cystic fibrosis
People with CF have a higher risk of developing certain other diseases, including cystic fibrosis-related diabetes (CFRD), colorectal cancer, and osteoporosis. Early detection is important to treat or manage these conditions. Your CF care team may recommend health screenings such as these:
Oral glucose tolerance test. Cystic fibrosis-related diabetes (CFRD) is one of the most common complications of CF in adults. If you have CF, you will likely be tested every year for CFRD, starting at age 10, with an oral glucose tolerance test (OGTT). The OGTT is the best way to diagnose CFRD and is usually done in the morning after an 8-hour fast. You will be asked to drink a “glucose drink” and then your blood glucose (sugar) will be measured at different times.
Colonoscopy. The risk for colorectal cancer in adults with cystic fibrosis is five to ten times greater than the general population, and even higher (20 times) for people with CF who receive a lung or other solid organ transplant. Because of this risk, it’s recommended that people with CF begin screening for colorectal cancer with a colonoscopy at age 40 (age 30 if you’ve had a solid organ transplant).
Dual-energy X-ray absorptiometry (DEXA) scan. People with CF are at risk for two common bone diseases: osteoporosis and osteopenia. These conditions can make your bones weak and brittle. Your CF care team will track your growth through height and weight, follow your development in puberty, and check your blood for vitamin D levels. It’s recommended that people with CF have a DEXA scan by age 18 and repeat the scan every 1-5 years. A DEXA scan is a type of X-ray that checks the thickness of your bones.
Takeaways
Cystic fibrosis (CF) is a genetic disease that affects the lungs, pancreas, and other organs. Although CF is a severe condition that needs daily care, there are many ways to treat it, and there’s been a great improvement in those treatments over the years. People who have CF now can expect to live a much longer life than those who had it in the past.
Cystic Fibrosis FAQs
What is the life expectancy of someone with cystic fibrosis?
People with CF continue to live longer and healthier lives. The Cystic Fibrosis Foundation Patient Registry collects data from patients who receive care for CF at CF Foundation-accredited care centers and have agreed to share their health information. Based on the 2022 Registry data, the life expectancy of people with CF who were born between 2018 and 2022 is predicted to be 56 years. Data from the 2021 Registry show that half of the babies born in 2021 are predicted to live to age 65 or older. A study based on clinical trials of people with CF taking the newer triple-combination CFTR modulator medication predicted possible lifespans of over 71 years.
Can people with cystic fibrosis have a normal life?
Most people with CF live a normal daily life, with the challenge of fitting in daily medications, airway clearance therapy, and other treatments and medications. Children with CF go to school, have friends, enjoy hobbies, and can exercise and play sports. Many go to college, get married, and have families of their own.
What happens if you don’t treat cystic fibrosis?
People with CF have thick, sticky mucus that blocks the airways in their lungs, making it difficult for them to breathe and easier to get infections. Treatments for CF include medicines to thin the mucus and fight infections, and therapies to clear the mucus from airways. CF also affects digestion, which makes it harder to absorb nutrients from food. There are medicines to help with this.
If a person with CF doesn’t treat these symptoms, they will likely have frequent lung infections, difficulty breathing, and long-term damage to the lungs. They may become malnourished from lack of nutrients and lose weight, which also makes it harder to fight lung infections. Without treatment, CF can lead to respiratory failure, intestinal blockages, organ failure, and death.
At what age can you be diagnosed with cystic fibrosis?
Every state in the U.S. tests newborns for cystic fibrosis. Newborn screening is done during the first few days of a baby's life, using only a few drops of blood from the heel. Though most people are diagnosed with CF by the age of 2, some are diagnosed as adults.
Posted : 2024-08-26 09:03
Read more

Disclaimer
Every effort has been made to ensure that the information provided by Drugslib.com is accurate, up-to-date, and complete, but no guarantee is made to that effect. Drug information contained herein may be time sensitive. Drugslib.com information has been compiled for use by healthcare practitioners and consumers in the United States and therefore Drugslib.com does not warrant that uses outside of the United States are appropriate, unless specifically indicated otherwise. Drugslib.com's drug information does not endorse drugs, diagnose patients or recommend therapy. Drugslib.com's drug information is an informational resource designed to assist licensed healthcare practitioners in caring for their patients and/or to serve consumers viewing this service as a supplement to, and not a substitute for, the expertise, skill, knowledge and judgment of healthcare practitioners.
The absence of a warning for a given drug or drug combination in no way should be construed to indicate that the drug or drug combination is safe, effective or appropriate for any given patient. Drugslib.com does not assume any responsibility for any aspect of healthcare administered with the aid of information Drugslib.com provides. The information contained herein is not intended to cover all possible uses, directions, precautions, warnings, drug interactions, allergic reactions, or adverse effects. If you have questions about the drugs you are taking, check with your doctor, nurse or pharmacist.
Popular Keywords
- metformin obat apa
- alahan panjang
- glimepiride obat apa
- takikardia adalah
- erau ernie
- pradiabetes
- besar88
- atrofi adalah
- kutu anjing
- trakeostomi
- mayzent pi
- enbrel auto injector not working
- enbrel interactions
- lenvima life expectancy
- leqvio pi
- what is lenvima
- lenvima pi
- empagliflozin-linagliptin
- encourage foundation for enbrel
- qulipta drug interactions